| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://jh.elmerpub.com |
Review
Volume 14, Number 6, December 2025, pages 281-296

Primary Gastrointestinal B-Cell Lymphomas: A Clinicopathological Review
Ibrahim Elsharawia, b ![]() , Christopher Liwskia
, Christopher Liwskia ![]()
aDepartment of Hematological Pathology, Dalhousie University, Halifax, Nova Scotia,
Canada
bCorresponding Author: Ibrahim Elsharawi, Department of Pathology and
Laboratory Medicine, Division of Hematological Pathology, Mackenzie Building, QEII Health
Sciences Centre, Halifax, NS B3H 1V8, Canada
Manuscript submitted November 19, 2025, accepted December 18, 2025, published online December 30,
2025
Short title: Primary Gastrointestinal B-Cell Lymphomas
doi:
https://doi.org/10.14740/jh2157
| Abstract | ▴Top |
The gastrointestinal tract is the most frequent extranodal site for lymphomas. Primary gastrointestinal B-cell lymphomas represent a distinct group of lymphoproliferative disorders and are the most common type of gastrointestinal lymphomas. Their pathogenesis, clinicopathological characteristics, and prognosis may differ from their nodal or systemic counterparts, emphasizing the importance of accurately identifying them. This review incorporates some of the latest literature on common primary gastrointestinal B-cell lymphomas focusing on their unique pathogenic mechanisms, clinical features, and pathological findings. It also addresses the prognostic outcomes associated with these lymphomas and provides a brief overview of the available treatment options.
Keywords: Gastrointestinal B-cell lymphomas; Primary gastrointestinal B-cell lymphomas; Primary gastrointestinal lymphoma; Lymphoma; Gastric lymphoma; H. pylori; MALT lymphoma
| Introduction | ▴Top |
The gastrointestinal (GI) tract is a host to various hematopoietic malignancies, with some exclusively arising in the GI system and others secondary to systemic disease [1-4]. Among the different cell lineages, lymphomas emerge as the most significant hematopoietic malignancies that show the potential to involve the GI tract in isolation [1, 2]. This tendency is likely attributed to the lymphoid-rich environment of the GI system and the local inciting factors that play a role in the pathogenesis of some of these lymphomas (e.g., Helicobacter pylori (H. pylori) infection) [1].
Non-Hodgkin B-cell lymphomas usually occur in the lymph nodes, spleen, or bone marrow, with approximately 30% of cases occurring outside of these regions [5]. The prevalence of extranodal lymphomas has increased owing to radiological advances in detecting them early and raised awareness in recognizing them [6]. Common extranodal sites include the skin, GI system, and head and neck [6]. The GI tract is the most common extranodal site for lymphomas, comprising up to 20% of cases, and it accounts for up to 4% of all GI malignancies [7]. The diagnostic criteria for primary GI lymphomas have been described by Dawson’s criteria which include: 1) absence of peripheral lymphadenopathy at presentation; 2) no mediastinal lymphadenopathy; 3) normal total and differential white blood cell count; 4) predominance of bowel lesion at the time of laparotomy with lymphadenopathy only noted close by; and 5) no involvement by lymphoma in the liver and spleen [8]. The modified Lugano staging system is often applied to primary GI lymphomas, which separates diseases into stage I (confined to the GI tract, including a single primary lesion or multiple noncontiguous lesions), stage II (extension into the abdomen from the primary GI site with stage II1 indicating local nodal involvement and stage II2 indicating distant nodal involvement), stage IIE (penetrating the serosa to involve adjacent organs or tissues), and stage IV (disseminated extranodal involvement or supradiaphragmatic nodal involvement) [9]. This review focused on GI lymphomas that arise from and predominantly involve any section of the GI tract, rather than secondary involvement of B-cell lymphomas of nodal origin or originating from another extranodal site.
Primary GI non-Hodgkin B-cell lymphomas account for a significant proportion of all primary GI lymphomas (∼90% of cases), while T-cell lymphomas and Hodgkin lymphomas only account for a minuscule fraction [7]. They are a heterogenous and unique group of lymphoproliferative disorders and may exhibit unique disease biology and clinical characteristics that differ from their nodal or extranodal non-GI counterparts. The purpose of this review is to provide an overview of the notable B-cell lymphomas that primarily affect the GI tract, presenting the common trends noted in these lymphomas while also addressing some unique characteristics that are helpful in distinguishing them.
| Search Strategy and Selection of Literature | ▴Top |
Identifying resources for this review relied on searching for appropriate research articles in the National Center for Biotechnology Information (NCBI) PubMed database. Various search terms were used for each of the entities covered, typically with a “primary gastrointestinal” modifier in the search line. All attempts were made to include recently published literature to best reflect the current knowledge of primary GI B-cell lymphomas. Preference was given to studies within the last 10 years, with the majority of the references used having been published between 2015 and 2025. However, this time range was expanded for certain key articles and primary studies that were important for describing some of the entities. Article types included a combination of primary studies and other reviews. In instances where numerous studies/reviews presented similar findings relevant to this review, a curated selection of references was made to prevent redundancy. Emphasis was placed on studies with larger sample sizes and more robust and clinically significant results. Case reports were also used for less common entities where current evidence is otherwise relatively limited. All articles were either written or translated in English.
This review discusses the following primary GI lymphomas: follicular lymphoma (FL), mantle cell lymphoma (MCL), marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma), diffuse large B-cell lymphoma (DLBCL), Burkitt lymphoma, plasmablastic (PB) lymphoma, and post-transplant lymphoproliferative disorder (PTLD). The review concludes with a brief discussion on the clinical and diagnostic approach to GI lymphomas along with a brief overview of other B-cell lymphomas rarely occurring in the GI tract. Table 1 summarizes some of the key characteristics of the primary GI B-cell lymphomas.
 Click to view |
Table 1. Summarized Key Characteristics of
Gastrointestinal B-Cell Lymphomas |
| Follicular Lymphomas | ▴Top |
Characterized by neoplastic proliferation of follicular B-cell lymphocytes, GI FL usually occurs in the fifth or sixth decades of life and does not appear to show a sex predilection [5, 6]. Most cases of GI FLs occur in the small intestine [2, 10]. The fifth edition of the WHO classification describes duodenal-type FL (DFL) as a distinct entity that accounts for 4% of all GI lymphomas [2, 10]. It can present with multifocal lesions [7]. Often the jejunum or ileum are involved concurrently [2]. Some case series have also described primary GI FL in the stomach and large intestine [1].
Pathogenesis
The pathogenesis of DFL shows similarities to that of systemic FL with t(14:18) rearrangements and mutations in genes such as TNFRSF14, EZH2, and KMT2D with varying frequencies [1, 2]. Chronic antigenic stimulation has also been hypothesized as a contributing factor to the development of FL [10]. A few case reports have highlighted that eradication of H. pylori, at least in part, has helped in the regression of some FLs of the duodenum, although this has not been substantiated through larger studies [11, 12].
Clinical features
Primary FL of the GI tract is typically discovered incidentally, and patients are usually asymptomatic [2]. In contrast, patients with secondary involvement of the GI tract may present with systemic symptoms [10]. When symptoms are present in primary GI FLs, they are non-specific, including abdominal discomfort, nausea/vomiting, pain, bleeding, and obstruction [10]. Positron emission tomography/computed tomography (PET/CT) scan is valuable in detecting the lesions and determining the stage [10]. Most cases have localized disease, presenting at low stage, and with a relatively indolent clinical course [2]. Iwamuro et al described a correlation between elevated soluble interleukin-2 receptor (sIL-2R) levels and systemic involvement when evaluating the staging based on Lugano and Ann Arbor systems [10].
Pathology
Gross/macroscopic appearance
On endoscopy, these lesions can appear as grey, white granular lesions with variable sizes [2]. Enlarged and fused villi and dilated vasculature have also been described, although best seen on endoscopy [10, 13]. Iwamuro et al reported that the combination of opaque white spots, enlarged villi, and dilated vessels may help to distinguish FLs from other white lesions on the typical differential diagnosis by endoscopy, such as lymphangiectasia, adenomas, and duodenitis [10]. FL can occasionally present as polypoid lesions as well [14] (Fig. 1). Ulceration of primary GI FLs is relatively rare [10].
 Click for large image |
Figure 1. Representative pathology image of a gastrointestinal follicular lymphoma. (a) Low power view of the colon biopsy with a polypoid appearance due to the underlying lymphoid infiltrate. The neoplastic lymphocytes are arranged in a follicular/nodular architecture and are based in the lamina propria with focal extension into the mucosa and submucosa (H&E, × 4). (b) High power view showing a mixture of small elongated cleaved cells (centrocytes) and large round/oval cells with prominent nucleoli (centroblasts) (H&E, × 40). The neoplastic lymphocytes show co-expression of both BCL6 (c, × 4) and BCL2 (d, × 4). H&E: hematoxylin and eosin. |
Histopathology
It typically presents with a follicular/nodular pattern of lymphoid infiltration commonly affecting the mucosa but may extend into the submucosa [2]. Resembling that of systemic low-grade FL, the lymphoid infiltrate is predominantly composed of a mixture of small to medium sized centrocytes and larger centroblasts (Fig. 1) [2]. Up to 95% of cases present as grade 1 or 2 FLs [10]. Occasionally, GI FLs present with ulcerated lesions hindering adequate morphological evaluation and leading to misdiagnosis and the need for repeated sampling [10]. Transformation to DLBCL is uncommon [10].
Immunohistochemistry
The immunophenotype usually mirrors that of systemic FL with positive B-cell markers (e.g., CD19, CD20, CD79a) as well as positive CD10, BCL2, and BCL6 [2, 10]. Utilizing follicular dendritic markers (e.g., CD21, CD23) is helpful in identifying a meshwork [2]. In DFL, the dendritic meshwork is typically pushed to the periphery, while other GI FLs have well preserved meshwork similar to that of nodal type [2]. H. pylori testing for upper GI FLs can be helpful to identify if further antibiotic therapy is warranted [12].
Prognosis/treatment
The prognosis of primary GI FL is generally favorable with most cases presenting with stage 1 disease [1, 2, 11, 14]. A subset of patients (up to 25%) undergo spontaneous regression [10]. Transformation to large cell lymphoma carries a worse prognosis, but transformation is rather rare in comparison to systemic FLs [10]. Studies have shown no disease-related mortality over a 10-year follow-up period [13]. Primary duodenal involvement appears to carry a better prognosis than FLs involving other parts of the small intestine [2]. Given that primary GI FLs tend to have an indolent clinical course, a watch and wait strategy is becoming popular as the main initial management approach [14, 15]. Alternative treatment modalities include chemotherapy with rituximab and bendamustine, immunotherapy, and radiotherapy for local control [10, 15, 16]. Since most studies are retrospective, optimal management of these cases continues to evolve. Historically, various treatment regimens were utilized including R-CHOP (rituximab-cyclophosphamide- doxorubicin-vincristine-prednisone), R-CVP (rituximab-cyclophosphamide-vincristine prednisone), CHVP-IFN (cyclophosphamide-doxorubicin-vindesine-prednisone-interferon), cyclophosphamide, and AVmCP (doxorubicin-teniposide-cyclophosphamide-prednisone) [14].
| Mantle Cell Lymphoma | ▴Top |
Primary GI MCL is an uncommon entity accounting for ∼2.8% of all newly diagnosed MCL cases based on a retrospective study by Castillino et al evaluating 800 cases [17]. Secondary involvement of the GI tract is much more common, accounting for almost 10% of newly diagnosed MCLs [17]. Overall, they represent up to 13% of all primary GI lymphomas [1]. Similar to MCL arising from lymph nodes or other extranodal sites, primary GI MCL typically arises in elderly individuals with a male predilection [17].
Pathogenesis
The pathogenesis of primary GI MCL is believed to mirror that of systemic or nodal MCL. The translocation of t(11;14) (q13;q32) which involves the IGH and CCND1 genes is largely recognized to be the key genetic event that ultimately leads to cyclin D1 overexpression as well as cell cycle dysregulation [2, 17].
Clinical features
The incidence of primary GI MCLs increases with age [7, 18]. The clinical presentation is highly variable, ranging from abdominal pain, GI bleeding, perforation, and obstructive symptoms [2, 17, 18]. Erosion and ulceration can rarely occur in primary GI MCLs, although it is more frequent in secondary GI MCLs [17]. Lower GI tract involvement is more common than upper GI tract, with the same trend observed in secondary involvement of the GI tract by systemic MCL [17]. Occasional primary involvement of the stomach has been described [18, 19]. The WHO highlights the role of FDG PET scans in diagnosis and prognostic evaluation, including staging, but also notes their limitations, underscoring the need for endoscopy, especially in patients with GI symptoms [2].
Pathology
Gross/macroscopic appearance
They can manifest as solitary lesions or multiple polyps involving long segments of the GI tract, also known as multiple lymphomatous polyposis [1, 2, 17]. The latter appears to be more common [17]. They can also present with large masses, superficial ulceration, or diffuse thickening of the mucosa [2].
Histopathology
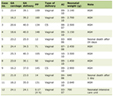
Microscopic evaluation typically reveals nodular lymphoid infiltrates involving the lamina propria and submucosa (Fig. 2). Diffuse involvement has also been described [18]. Lymphocytes are typically small to medium sized with irregular nuclear contours [2, 19]. Blastoid, an aggressive histological variant, has also been reported in primary GI MCLs [2, 17, 18]. The small cell variant has also been reported which can be confused for chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), highlighting the essential role of cyclin D1 in confirming the MCL diagnosis [2]. The frequency of the histologic variants between primary GI MCL and secondary MCL of the GI tract is similar [17].
 Click for large image |
Figure 2. Representative case of a primary gastrointestinal mantle cell lymphoma. (a) Low power view shows a nodular lymphocyte infiltrate in the lamina propria of a colon biopsy (H&E, × 4). (b) The neoplastic lymphocytes are small to medium sized with angulated nuclei (H&E, × 40). These cells are positive for CD5 (c, × 20) and cyclin D1 (d, × 20). H&E: hematoxylin and eosin. |
Immunohistochemistry/ancillary studies
In addition to B-cell markers, MCL is typically positive for CD5, cyclin D1, and SOX11, and negative for germinal center markers (CD10 and BCL6) (Fig. 2) [2, 7]. Molecular detection of t(11;14) (q13;q32) can be helpful in confirming the diagnosis if needed [2, 18, 19]. TP53 testing by next generation sequencing or immunohistochemistry can be helpful in further risk stratification [2, 20].
Prognosis/treatment
Although patients with primary GI MCLs appear to show relatively lower MCL International Prognostic Index (MIPI) scores, their overall outcomes are similar to that of secondary GI MCLs [17]. The prognosis overall appears to be favorable with > 70% of cases having a low MIPI score [17]. The overall survival and progression-free survival do not appear to be impacted by the number of lesions at presentation [17]. With regard to treatment, a tailored approach based on the extent of GI involvement should be considered. Conservative management by observation and/or localized therapy can be taken if the GI involvement is limited [17]. Rituximab monotherapy, R-CHOP, R-CVP, and autologous-hematopoietic stem cell transplant (HSCT) have been traditionally used, especially in patients with multiple lesions or extensive GI involvement [1, 17, 18, 21]. The use of novel agents such as Bruton’s tyrosine kinase (BTK) inhibitors (e.g., ibrutinib), venetoclax (BCL2-inhibitor), and immunomodulatory drug such as lenalidomide are gaining traction, although further research is warranted to establish their role in primary GI MCL [17, 20]. Chimeric antigen receptor (CAR) T-cell therapy may also play a role in the future [17]. According to Castellino et al, primary GI MCL is typically treated with less aggressive modalities in comparison to patients with secondary involvement of the GI tract [17]. Owing to the recent advances in therapeutic approaches, there is improved overall survival, particularly in younger patients who can undergo chemo-immunotherapy followed by stem cell transplant [18, 21]. For older patients who are not fit for transplant, the use of rituximab for remission induction can improve outcomes in these patients [18, 21].
| MALT Lymphoma | ▴Top |
MALT lymphoma is the most common type of marginal zone lymphoma [22]. They account for 5-8% of B-cell lymphomas and can affect any tissue in the body [2, 22]. The incidence of MALT lymphomas has increased over the years except for gastric MALT lymphomas, likely due to the effective treatment of H. pylori [2, 22, 23]. The most common location for MALT lymphomas is the stomach accounting for up to 35% of all cases [23]. Gastric MALT lymphomas are also the most common type of gastric lymphomas (up to 60%) [2, 24]. Colorectal and small intestinal MALT lymphomas are less common, comprising 5.2% and 3.4% of MALT lymphoma cases, respectively [7, 22, 23]. Multifocal and concurrent involvement of both the stomach and intestines can occur rarely [7, 23]. Large intestinal MALT lymphomas tend to have a female predilection [23].
Pathogenesis
The pathogenesis of MALT lymphomas are heterogenous. The primary process involves an immune response dysregulation with somatic hypermutations in clonally rearranged immunoglobulin genes [2, 22]. Translocation of chromosomes 11 and 18 is relatively common, accounting for up to 24% of gastric MALT lymphomas, and it also represents the most common translocation in H. pylori-negative MALT lymphomas [2, 21, 23]. The association between H. pylori and gastric MALT lymphomas has been well established with some literature also highlighting the relationship between this organism and colorectal MALT lymphomas in about 20% of cases [2, 23, 24]. Campylobacter jejuni infection has been linked to small intestinal MALT lymphoma [22, 23]. Immunoproliferative small intestinal disease/alpha heavy chains (IPSID/αHCD), a subtype of GI MALT lymphoma with dysfunctional immunoglobulin alpha chains, have also been variably associated with Campylobacter jejuni and usually occur in younger patients living in low socioeconomic conditions [1, 2, 5, 23].
Clinical presentation
Many patients with primary gastric involvement are asymptomatic at presentation while those with intestinal MALT lymphomas can present with abdominal pain and abnormal bowel habits (constipation and diarrhea) and GI bleeding [5, 23]. Mucosal ulceration can occasionally occur and can be identified on endoscopy [24, 25].
Pathology
Gross/macroscopic appearance
Similar to other primary GI lymphomas, MALT lymphomas can grossly appear as polypoid lesions, diffuse thickening of the stomach or intestinal wall, or large masses associated with ulceration [24, 26].
Histopathology
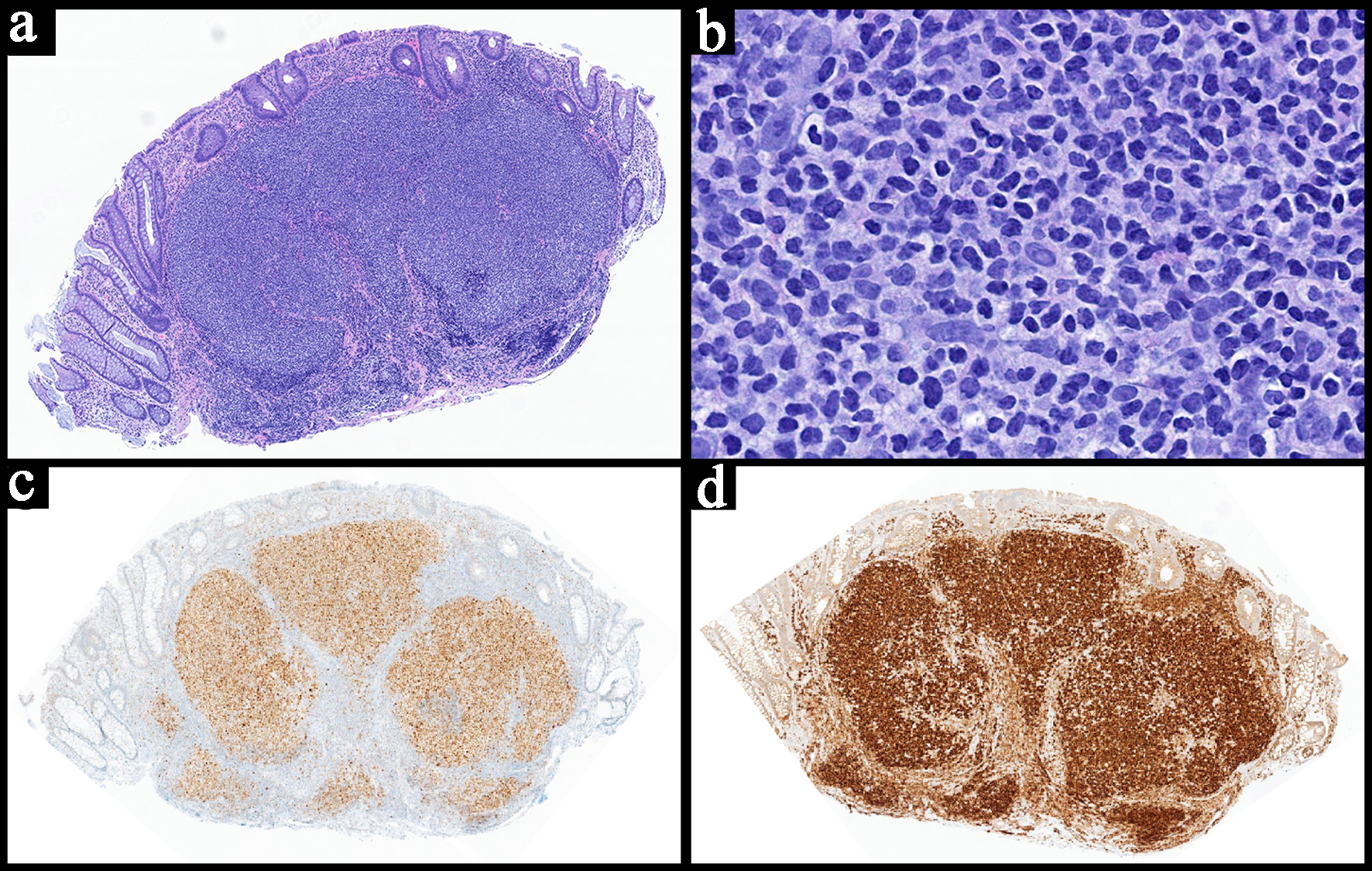
On histopathological evaluation, primary GI MALT lymphomas present with sheets of neoplastic small lymphocytes with angulated nuclei and monocytoid appearance with abundant cytoplasm (Fig. 3). They tend to infiltrate around reactive lymphoid follicles, which can make it difficult to distinguish from reactive lymphoid hyperplasia at times [2]. Plasmacytic differentiation is relatively common, occurring in approximately 33% of cases [23]. The IPSID/αHCD variant typically presents with extensive plasma cell differentiation [2, 23]. Occasional transformed lymphocytes may be seen with centroblastic and immunoblastic morphology. Neoplastic lymphocytes are often seen replacing the germinal centers, a phenomenon known as follicular colonization [2, 26]. The infiltration of the glandular epithelium forming lymphoepithelial lesions is specific in establishing a pathological diagnosis, although it lacks sensitivity (Fig. 3) [21, 26]. Eosinophilic degeneration of the epithelium may be seen in lymphoepithelial lesions, indicating chronic damage [26]. Amyloid deposition can be seen and H. pylori organisms can be noted by hematoxylin and eosin (H&E) staining if they are abundant [26].
 Click for large image |
Figure 3. Representative case for extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue in the stomach. (a) Low power view showing a diffuse lymphoid infiltrate in the mucosa, submucosa and lamina propria (H&E, × 4). (b) High power view of the neoplastic lymphocytes that appear small to medium sized with some showing a monocytoid morphology. The neoplastic lymphocytes are noted infiltrating in and around the glands forming lymphoepithelial lesions (H&E, × 40). (c) CD20 highlights the neoplastic lymphocytes confirming their B-cell lineage (× 5). (d) H. pylori immunohistochemical stain is positive (red arrow) (× 40). Molecular studies for B-cell clonality were also positive. H&E: hematoxylin and eosin. |
Immunohistochemistry/ancillary studies
The neoplastic lymphocytes are positive for B-cell markers (CD19, CD20, CD79a) and occasionally BCL2 (Fig. 3) [2, 26]. They are negative for CD5 and CD10, distinguishing them from MCL and FL. Kappa or lambda restriction is helpful for establishing clonality and are often done by flow cytometry [26].
Given that these lesions lack specific immunophotypical aberrancies in comparison to other B-cell lymphomas (e.g., aberrant CD5 expression in MCL) and their potential overlap with reactive conditions, MALT lymphomas can be difficult to diagnose and might require repeated follow up specimens. B-cell clonality studies can play an important role when flow cytometry is not available to confirm clonality [26]. In cases where there is extensive lymphoplasmacytic differentiation, MYD88 testing to rule out lymphoplasmacytic lymphoma (LPL) may be recommended, although primary GI involvement by LPL is quite rare [2, 27]. Finally, testing for the presence of H. pylori, either by special stains or immunohistochemistry, is an important consideration, especially in gastric MALT lymphomas (Fig. 3) [2, 26].
Prognosis/treatment
GI MALT lymphomas usually have an indolent clinical course with only 2% of cases eventually progressing to an aggressive B-cell lymphoma [23]. The 5-year survival is up to 90% [2]. Patients with stomach MALT lymphomas have a sixfold increase in developing a gastric carcinoma [23, 24]. However, stomach MALT lymphomas have a better prognosis than their intestinal counterparts, likely due to the well-defined management guidelines established for their treatment [21, 24].
Eradication of H. pylori can resolve gastric MALT lymphomas in about 73-75% of cases [22, 24]. However, recurrence can occur post eradication therapy in 3-21% of cases [23]. Interestingly, antibiotic therapy for colorectal MALT lymphomas has been shown to be effective, but the exact mechanism is yet to be studied [23]. A positive translocation t(11;18) and t(1;14) status, deep infiltration of the tumor, and H. pylori negativity are associated with resistance to antibiotic treatment [22, 23, 25]. Interval follow-up and surveillance starting 3 months after therapy completion is recommended for gastric MALT lymphomas to ensure resolution [21]. If eradication therapy fails, several management options have been described including watch and wait, local surgical resection, radiotherapy, and immunochemotherapy [5, 23, 24]. Various agents have been used with rituximab and alkylating agents (e.g., bendamustine) being the most common [23, 24]. A single agent or multi-agent approach can be taken based on clinical and patient factors [22, 23]. There are no definitive guidelines on managing non-gastric GI MALT lymphomas, and the literature has highlighted that remission rates are relatively low with H. pylori eradication therapy alone [7, 22].
| Primary GI DLBCL | ▴Top |
Primary GI DLBCL is the most common subtype of primary GI lymphoma [2, 28]. Recent studies have focused on examining GI DLBCLs based on their location, whether it is the stomach or the intestines [28-32]. The commonest site of involvement is the stomach, with DLBCL of the body being the most frequent (57%), followed by the antrum (51%), fundus (14%), and cardia (3%) [31]. The majority of primary intestinal DLBCLs arise from the ileocecal region (50%), with the jejunem and ileum being the next most common sites (23%) [33]. DLBCL of the duodenum comprises 18% of cases and is second in frequency to FL [33]. Colonic and rectal involvement are less common. DLBCL of the esophagus has been rarely reported [29, 30]. In general, primary GI DLBCL has a male predilection and typically presents in the fifth to sixth decade of life [2, 28].
Pathogenesis
The exact etiology for primary GI DLBCL is not entirely clear. Some theories suggest that inflammation of the GI tract and overstimulation of lymphocytes in the MALT promotes disease development [29, 31]. In particular, H. pylori infection has been associated with gastric DLBCL [29, 31]. Epstein-Barr virus (EBV) infection, immunodeficiency, inflammatory bowel disease, genetic factors, and medications have also been postulated to contribute to the neoplastic transformation [2, 29]. Genetic aberrations, including MYC, BCL2, and BCL6 rearrangements, TP53 mutations, alterations in the nuclear factor-κB (NF-κB) pathway, and B-cell receptor signaling have been identified and are generally similar to DLBCLs in other sites [2, 34]. Some site-specific differences include lower rates of MYD88 and PIM1 mutations in primary GI DLBCL compared to other extranodal types, such as DLBCL of the breast and testes [34].
Clinical presentation
The clinical manifestations of primary GI DLBCL can include nausea/vomiting, indigestion, loss of appetite, abdominal pain, bleeding, obstruction, and perforation [2, 29, 30]. The findings are non-specific, potentially delaying diagnosis [29, 30]. Similarly, endoscopic findings may range from minimal irregularities to ulceration [30, 33]. The presence of auriculate ulcerative mounds is thought to be characteristic of DLBCL arising in the duodenum [33].
Pathology
Gross/macroscopic appearance
The tumors are often visible as fleshy masses associated with mucosal ulceration. Thickening and transmural involvement are typically evident [2, 33].
Histopathology
Histopathological findings are typically similar to DLBCLs in other sites, presenting as a proliferation of atypical, medium-to-large-sized lymphoid cells arranged in sheets (Fig. 4) [2, 35]. The morphology is usually centroblastic or immunoblastic [2]. Frequent mitotic figures, apoptotic debris, and areas of necrosis may be observed [31]. Transmural involvement is common (Fig. 4) [2, 29]. In some cases, a background low-grade component may be identified, including FL and MALT lymphoma [2, 31, 36]. These background components may be more frequently observed in the stomach, typically from MALT lymphoma, and in the duodenum where transformation from FL has been occasionally seen [29, 33, 36]. In contrast, disease involving the ileocecal region typically arises de novo [28, 33, 36].
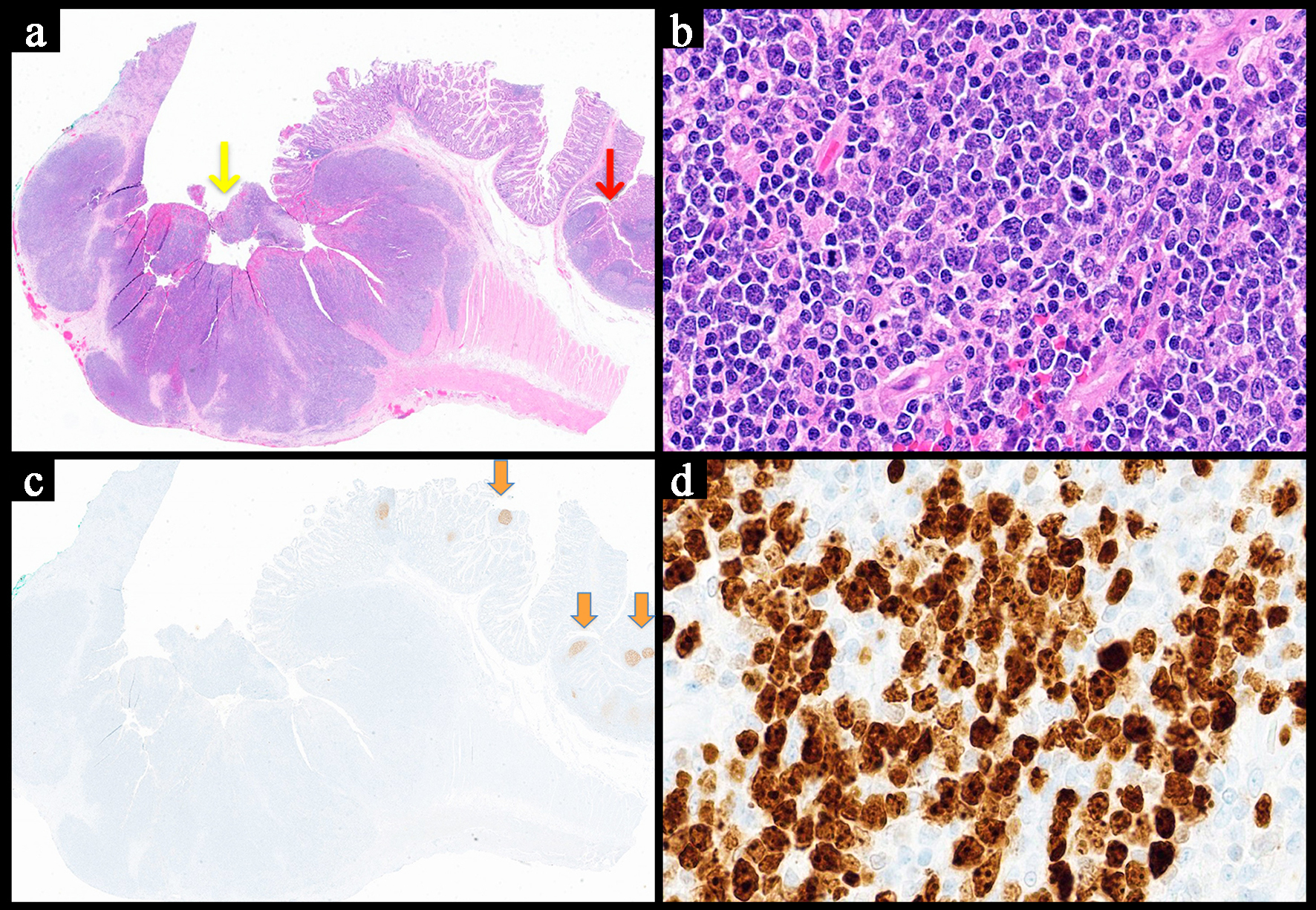 Click for large image |
Figure 4. Representative image of a diffuse large B-cell lymphoma involving the small bowel. (a) Low power view showing sheets of neoplastic lymphocytes with full thickness involvement of the bowel wall and surface ulceration of the mucosa (yellow arrow). A few normal appearing lymphoid follicular are noted at the right edge (red arrows) (H&E, × 1). (b) The cells are growing in sheets and appear large and pleomorphic with scattered atypical mitotic figures (H&E, × 40). (c) CD21 stain highlighting the lack of follicular dendritic meshwork and intact meshwork (orange arrow) in the benign lymphoid follicles (acting as an internal control) (× 1). (d) The Ki-67 proliferative index is high reaching around 70% (× 40). H&E: hematoxylin and eosin. |
Immunohistochemistry/ancillary studies
The neoplastic cells express one or more B-cell markers (CD19, CD20, CD79a, PAX5) [2]. Both germinal center B (GCB) phenotype (CD10 positive and/or BCL6 positivity without IRF4 (MUM1) expression) and non-GCB phenotype (CD10 negative with strong IRF4) have been observed and are estimated to occur with approximately equal frequencies [2, 31, 36]. The absence of follicular dendritic meshwork can be highlighted by CD21 and CD23 stains (Fig. 4) [2]. The Ki-67 index is high (> 40%) [2, 35]. In situ hybridization for EBV-encoded RNA (EBER) can highlight infected cells in cases associated with EBV [2, 29]. However, according to the fifth edition of the WHO classification, such cases should be classified as EBV-positive DLBCL, not otherwise specified (NOS) [2]. In cases involving the stomach, H. pylori immunohistochemical stains can help to highlight the bacteria [28, 31].
Prognosis/treatment
Generally, patients with primary GI DLBCL have a more favorable prognosis than nodal subtypes and systemic subtypes with other extranodal sites of involvement, including the bone marrow, kidney/adrenal glands, central nervous system (CNS), and skin [2, 31, 34]. Multimodal therapy can be used in the treatment of GI DLBCLs, including surgical resection and chemoimmunotherapy with or without radiotherapy [28, 29, 31]. The chemoimmunotherapy protocols are similar to those in nodal DLBCL types, typically involving treatment with R-CHOP [29]. More recently, immune checkpoint inhibitors and CAR T cells have been employed [28]. Surgical resection can help reduce perforations, obstruction, and bleeding complications [28, 29, 31]. However, in gastric DLBCLs, total gastrectomy has been associated with serious post-operative complications and poor quality of life, with systemic therapies generally preferred [28]. A patient’s H. pylori status may also influence the treatment for gastric DLBCL. H. pylori infection is associated with improved overall survival and sensitivity to chemotherapy [28]. H. pylori eradication does not appear to have a significant benefit in addition to chemoimmunotherapy but is recommended by some authors due to its low probability of causing harm to the patient and potentially being able to mitigate the known risk for developing adenocarcinoma in H. pylori-infected individuals [35]. In contrast, EBV infection confers a poor prognosis and is associated with higher rates of refractory and relapsed disease [29, 36]. Additional poor prognostic factors include perforation, age ≥ 65, advanced stage, non-GCB type, programmed death-ligand 1 (PD-L1) expression, double expressor status, TP53 mutation, CDKN2A deletion, and trisomy 3 [2, 29].
| Burkitt Lymphoma | ▴Top |
Burkitt lymphoma is relatively rare compared to other primary GI B-cell lymphomas, representing approximately 4-5% of cases within this category [37, 38]. Of the three main clinical subtypes of Burkitt lymphoma, the sporadic type most commonly involves the GI tract, particularly the ileocecal area [38, 39]. The small intestine is the next most frequent GI site of involvement, while gastric and colonic Burkitt lymphomas are rare [5, 40, 41]. In general, Burkitt lymphomas are most commonly seen in children and young adults with a male predilection [2, 42]. However, based on a study of American Surveillance, Epidemiology, and End Results (SEER) data, primary GI Burkitt lymphomas are more commonly seen in older individuals, with a median age of 47 [38]. The same study also showed that the frequency of elderly individuals (defined as an age ≥ 60 years) with primary gastric Burkitt lymphoma was higher compared to the intestinal type [38].
Pathogenesis
Burkitt lymphoma can be subclassified into endemic, sporadic, and immunodeficiency-associated types. EBV infection is a strong etiological factor in disease development, showing a particularly high association in the endemic type (> 95%), with relatively lower frequencies in the sporadic (20-30%) and immunodeficiency-associated (25-40%) types [2, 39]. The endemic type is seen in tropical Africa with plasmodium falciparum infection also demonstrating a strong association [2, 21]. The predominant genetic aberrations that drive Burkitt lymphoma pathogenesis are translocations of the MYC gene, most commonly to the immunoglobulin heavy chain (IGH) locus (t(8;14)(q24;q32)) [2].
Clinical presentation
Burkitt lymphoma is a high-grade, aggressive lymphoma. Due to its high proliferation rate, masses caused by the disease may rapidly increase in size and produce corresponding symptoms. Involvement of the GI tract often manifests as abdominal pain, nausea/vomiting, palpable mass, weight loss, obstruction, bleeding, and intussusception [2, 40, 41].
Pathology
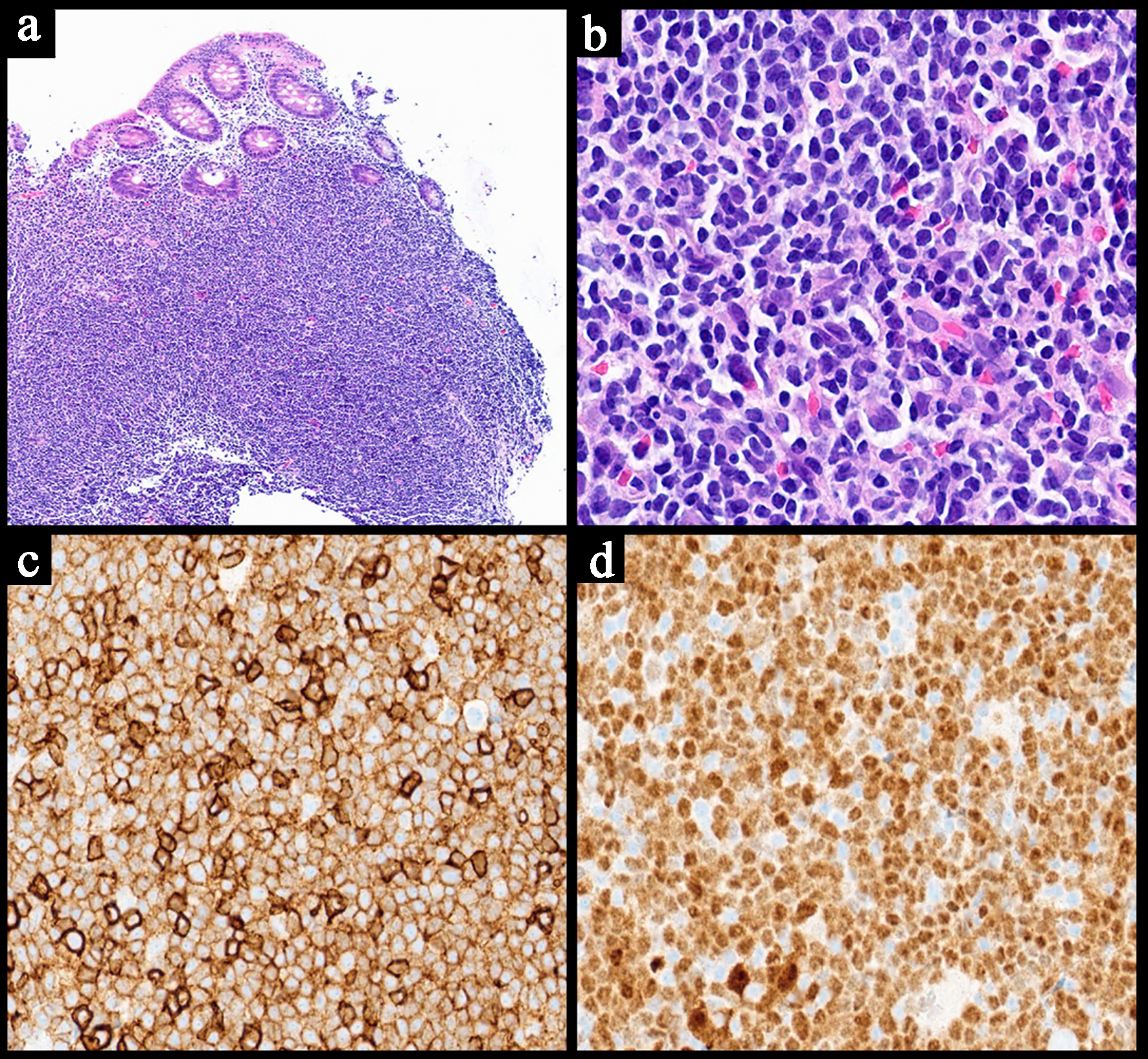
Gross/macroscopic appearance
According to the WHO, Burkitt lymphomas macroscopically present as soft, tan fleshy tumors with hemorrhage and necrosis [2]. The masses may show central ulceration (Fig. 5) [42].
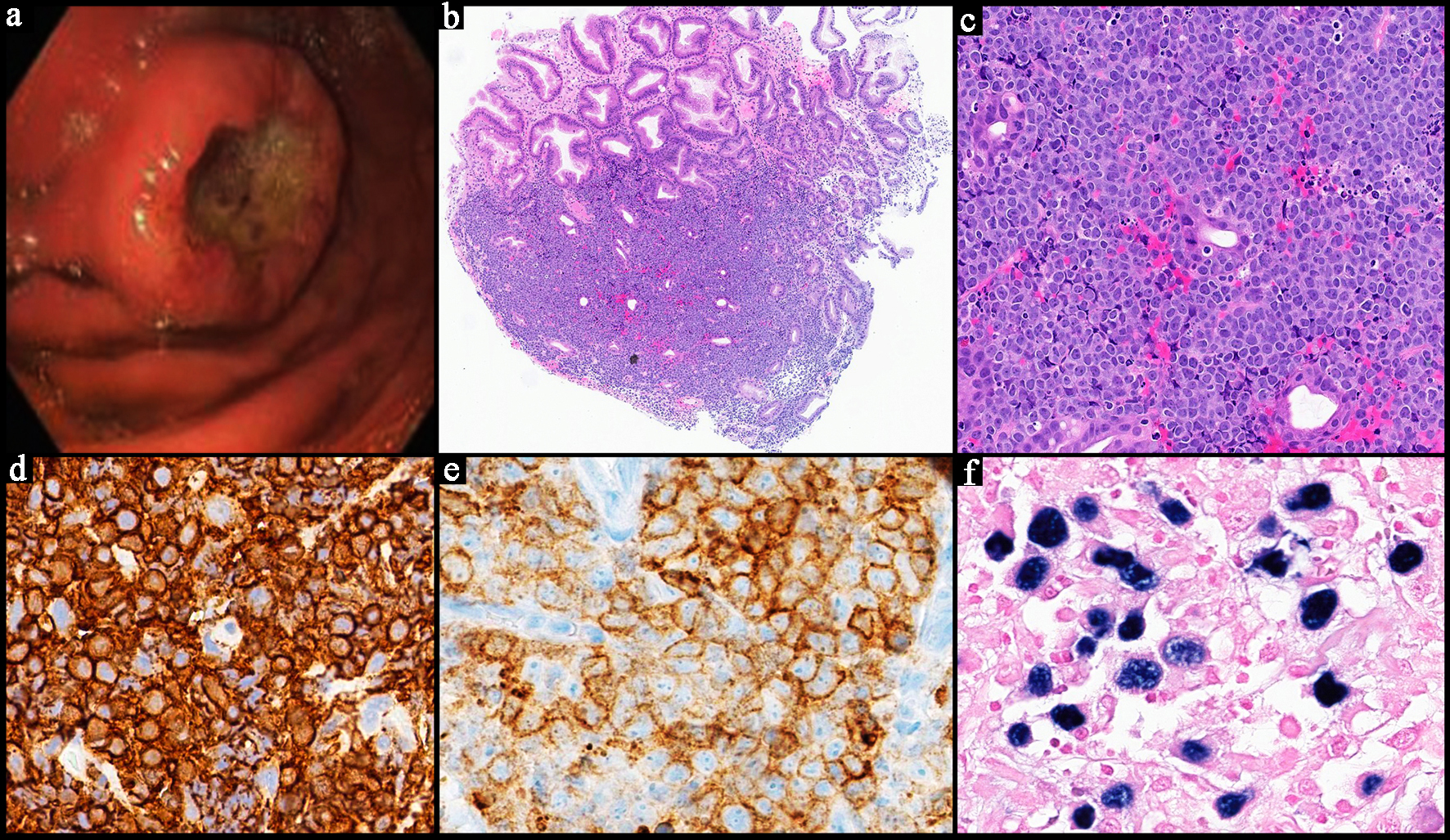 Click for large image |
Figure 5. Representative case of a Burkitt lymphoma involving the stomach. (a) The endoscopic image of the stomach mass with central ulceration. (b) Low power view of a neoplastic lymphoid proliferation in sheets involving the mucosa of the stomach and infiltrating around the glands (H&E, × 4). (c) Higher power image of medium sized neoplastic lymphocytes with cytological atypia and scattered apoptotic bodies showing a “starry sky” appearance (H&E, × 20). The neoplastic cells are positive for CD20 (d, × 40), CD10 (e, × 40) and Epstein-Barr virus-encoded RNA (EBER) in situ hybridization (f, × 40). H&E: hematoxylin and eosin. |
Histopathology
Histologically, Burkitt lymphomas can be classically recognized by a “starry sky” pattern produced by scattered tingible body macrophages with apoptotic debris admixed within a diffuse proliferation of lymphoma cells (Fig. 5) [2, 39]. When it occurs in the stomach, they can be seen infiltrating around gastric glands (Fig. 5) [42]. The neoplastic cells are typically medium-sized with round nuclei, multiple paracentral nucleoli, basophilic cytoplasm (occasionally with vacuoles), and squared off cell borders [2, 21, 39]. Owing to their high proliferation rate, frequent mitotic figures are often visible [2, 39].
Immunohistochemistry/ancillary studies
The neoplastic cells of Burkitt lymphoma are typically positive for CD19, CD20, CD79a, PAX5, CD10, BCL6, and c-MYC, with the latter showing particularly strong expression (Fig. 5) [2, 39]. The Ki-67 index is invariably high, often approaching 100% expression [2, 39]. CD5, CD23, CD138, BCL2, and TdT are classically negative [2, 39]. The presence of MYC gene translocations can be demonstrated by molecular testing [2, 41].
Prognosis/treatment
Burkitt lymphomas typically respond well to multi-agent chemotherapy, often including a combination of cyclophosphamide, prednisolone, and vincristine with or without methotrexate and cytarabine [5, 39]. HSCT could be valuable post HSCT [7]. Surgery also plays an important role in cytoreduction and reducing complications related to abdominal masses and intussusception [40, 43]. The overall prognosis is poor but appears to be improving with recent therapeutic advances [1, 21, 42]. For primary GI Burkitt lymphomas, the site of involvement appears to influence overall survival. According to Xie et al, patients with gastric Burkitt lymphoma have poorer overall survival compared to patients with primary intestinal disease [38].
| Plasmablastic Lymphoma | ▴Top |
GI PB lymphoma is a distinct entity described in the WHO (hematolymphoid tumors of the digestive system) [2]. It is a rare condition occurring in various nodal and extranodal sites [2, 44]. Primary GI involvement makes up approximately 20% of all extranodal PB lymphomas, and the available literature on it is relatively limited [2, 44].
Pathogenesis
Several pathogenic mechanisms have been described in PB lymphomas including immunoglobulin heavy chain variable region (IGHV) rearrangements, MYC translocations with immunoglobulin genes, and PRDM1/Blimp1 mutations. It can also be driven by EBV infection, especially in human immunodeficiency virus (HIV)-infected patients [2]. It has also been associated with autoimmune disorders and inflammatory bowel disease [45, 46].
Clinical presentation
They are typically more frequent in males than females [2, 45], and are associated with immune deficiency states, accounting for 2% of all HIV-related lymphomas [2, 45]. While they have historically been described in HIV-positive patients, further literature has shown that they can occur in HIV-negative patients in 31.1% of cases [25, 44, 45]. They can occur in any location in the GI tract, including the esophagus and anal canal [1, 2, 21, 45]. Colon involvement is relatively more common in HIV-negative cases [45]. They usually manifest as a local mass initially but may present with advanced stage in HIV-positive patients [2]. Clinical symptoms include GI manifestations such as diarrhea and abdominal pain as well as B-symptoms [45].
Pathology
They are typically characterized with diffuse infiltration of the lamina propria by neoplastic cells growing in sheets [2, 45]. These neoplastic cells are large and exhibit PB and immunoblastic morphology (Fig. 6) [2, 45]. A starry sky appearance similar to Burkitt lymphoma may also be seen (Fig. 6). The neoplasm commonly has brisk mitotic activity and can show areas of necrosis and granulomas [2, 45].
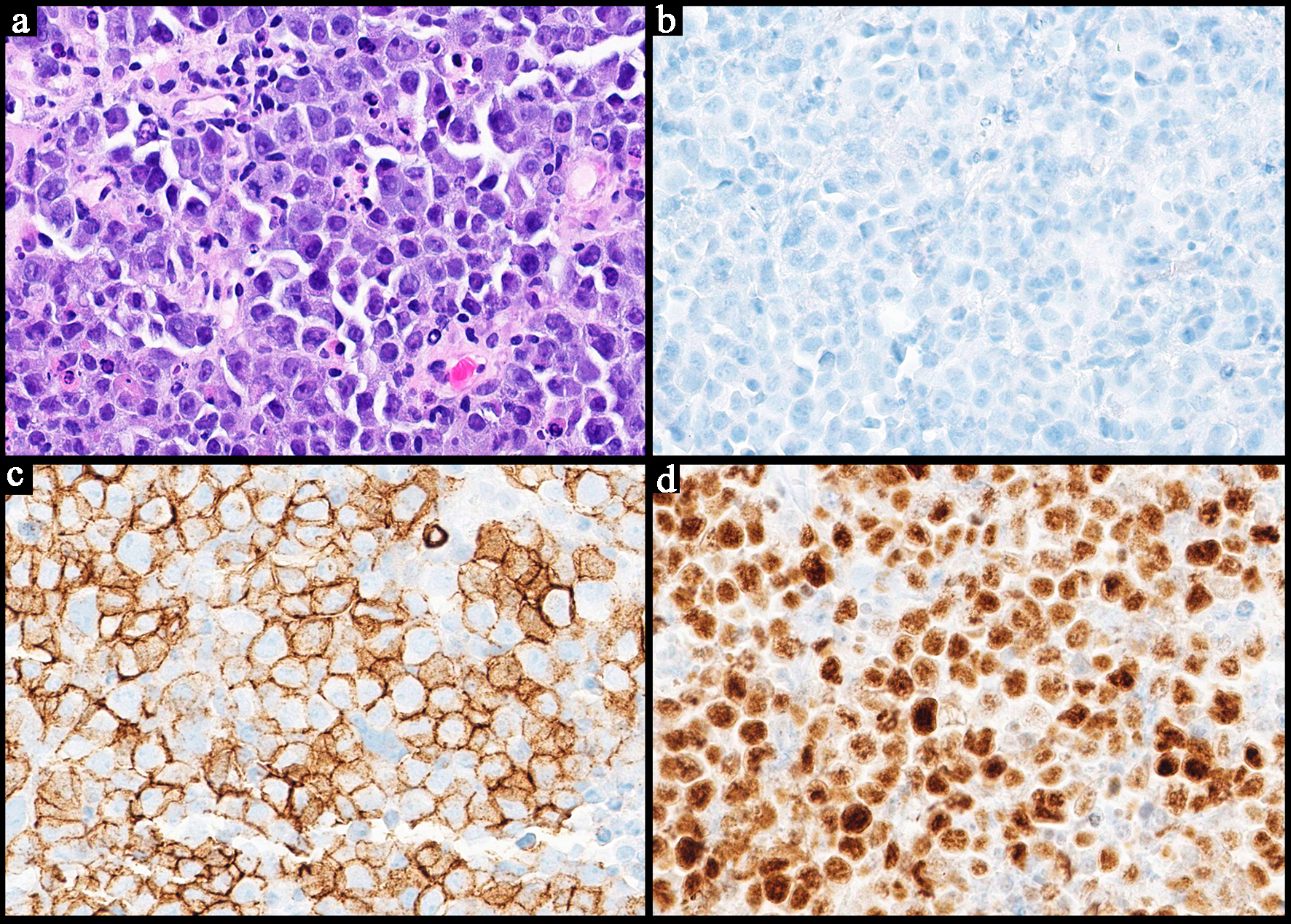 Click for large image |
Figure 6. Representative case of a plasmablastic lymphoma. (a) Highlights sheets of neoplastic proliferation of medium to large immunoblasts and plasmablasts (with eccentric nuclei). They are exhibiting cytotological atypia with prominent nucleoli. Foci of apoptotic debris are noted reminiscent of that seen in Burkitt lymphoma (H&E, × 40). The neoplastic cells are negative for CD20, a B-cell marker (b, × 40). They are positive for CD138, a plasma cell marker (c, × 40). They are also positive for c-MYC (d, × 40). H&E: hematoxylin and eosin. |
Contrary to the classic B-cell lymphomas described above, they are usually negative for CD20 and show weaker expression of other B-cell markers (e.g. PAX5, CD79a, CD45) (Fig. 5) [2]. Diagnosis can be challenging due to the lack of expression of these markers, their undifferentiated appearance, and their expression of EMA (an epithelial marker) [45]. A broad immunohistochemical panel may be warranted to establish a diagnosis. MUM1 and plasma cell markers such as CD138 and CD38 are typically positive (Fig. 5). Kappa and lambda stains are helpful in establishing clonality [2]. Ki-67 proliferative index is usually high (> 90%) and EBER is positive in 82% of HIV-positive cases and up to 60% in HIV-negative cases [1, 2]. Immunostain for c-MYC is usually positive (Fig. 5) [2].
Prognosis/treatment
PB lymphoma is an aggressive lymphoma with a mean survival of 9 - 15 months [1, 2]. An HIV-negative status and MYC translocation are associated with worse outcomes [2]. With regard to treatment, many patients end up receiving palliative care, but chemotherapy and HSCTs have been described as feasible therapeutic options. Examples of proposed chemotherapy regimens in the literature include etoposide-vincristine-doxorubicin-cyclophosphamide-prednisone (EPOCH) and cyclophosphamide-vincristine-doxorubicin-methotrexate alternating with ifosfamide, etoposide, and cytarabine (CODOX-M/IVAC) [44, 46, 47].
| PTLD | ▴Top |
PTLDs are uncommon lymphomas that occur in the context of a known history of solid organ transplantation or HSCT [2, 48, 49]. They are characterized by a heterogenous neoplastic lymphoid proliferation [2]. They arise in nodal and extranodal sites with the GI tract being the most common extranodal site (up to 30% of cases) [2, 21, 25, 48]. The cumulative incidence of PTLD post-transplant is 1.0±0.3% at 10 years [50]. Based on a review conducted by Reiche et al, the frequency of occurrence in various GI sites varies significantly based on different studies and clinical circumstances (e.g., type of transplant), but it appears that stomach and small intestines are the most common locations within the GI tract [48, 50-52].
Pathogenesis
Multiple factors are involved in the pathogenesis of PTLPDs. Notably, latent EBV infection plays a major role via various pathways including the activation of JAK/STAT, MAPK, and NF-κB pathways [2, 53]. It also upregulates antiapoptotic proteins such as BCL2, A20, and C-FLIP [53]. In addition, the immunosuppression coupled by EBV infection hinders the T-cell response [2, 53]. Hepatitis C and human herpesvirus 8 (HHV-8) infection also appear to be risk factors, especially when patients are seropositive for EBV [48].
Clinical presentation
Patients can present with nonspecific symptoms such as fever, mass effect, or GI complications such as bleeding, obstruction, and perforation [2, 49]. Endoscopic findings usually reveal rubbery, raised, erythematous lesions with central ulcers, circumferential ulcers, or nodular lesions with or without central ulceration [49].
Pathology
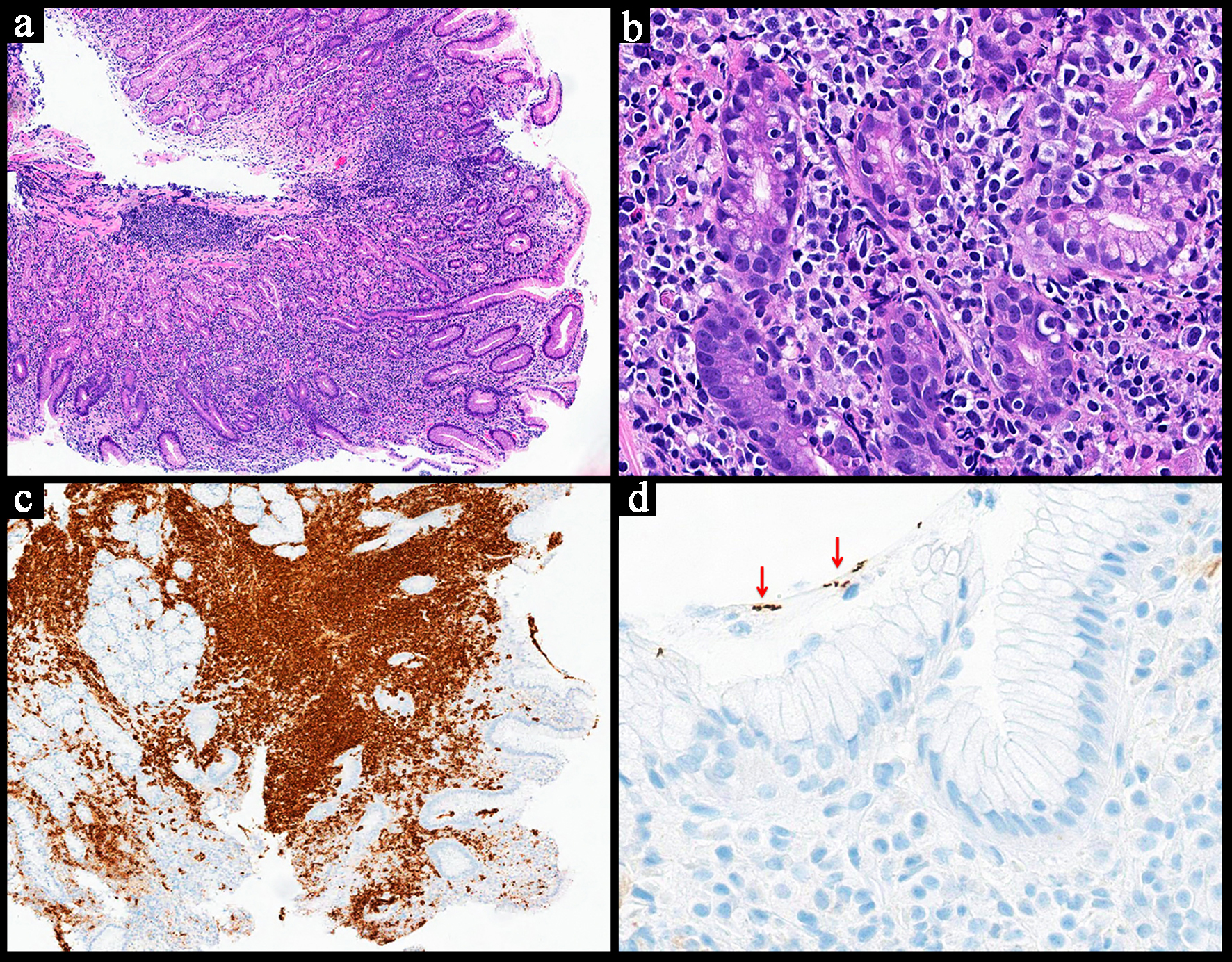
The fifth edition of the WHO classification categorizes PTLDs based on their clinicopathological features into non-destructive PTLD, polymorphic PTLD (P-PRLD), monomorphic PTLD (M-PTLD), classic Hodgkin lymphoma, and mucocutaneous ulcer [2, 21]. P-PTLD and M-PTLD appear to involve the GI tract most frequently [2]. P-PTLD usually manifests with polyclonal B cells admixed with T cells [2]. Clonal B cells can also be present [2]. M-PTLD shows monoclonal lymphocytes and appears similar to EBV+ DLBCL. The neoplastic lymphocytes can also be of T-cell or plasma cell lineage [2].
B-cell dominant PTLDs present with atypical lymphocytes infiltrating mucosa and into deeper levels, sometimes involving serosa (Fig. 7) [49]. They are positive for B-cell markers (e.g., CD20), negative for T-cell markers (e.g. CD3), and are usually EBER-positive (Fig. 7) [2, 49]. For polymorphic cases, there will be a heterogenous population of B and T cells, each expressing their specific markers [2, 54].
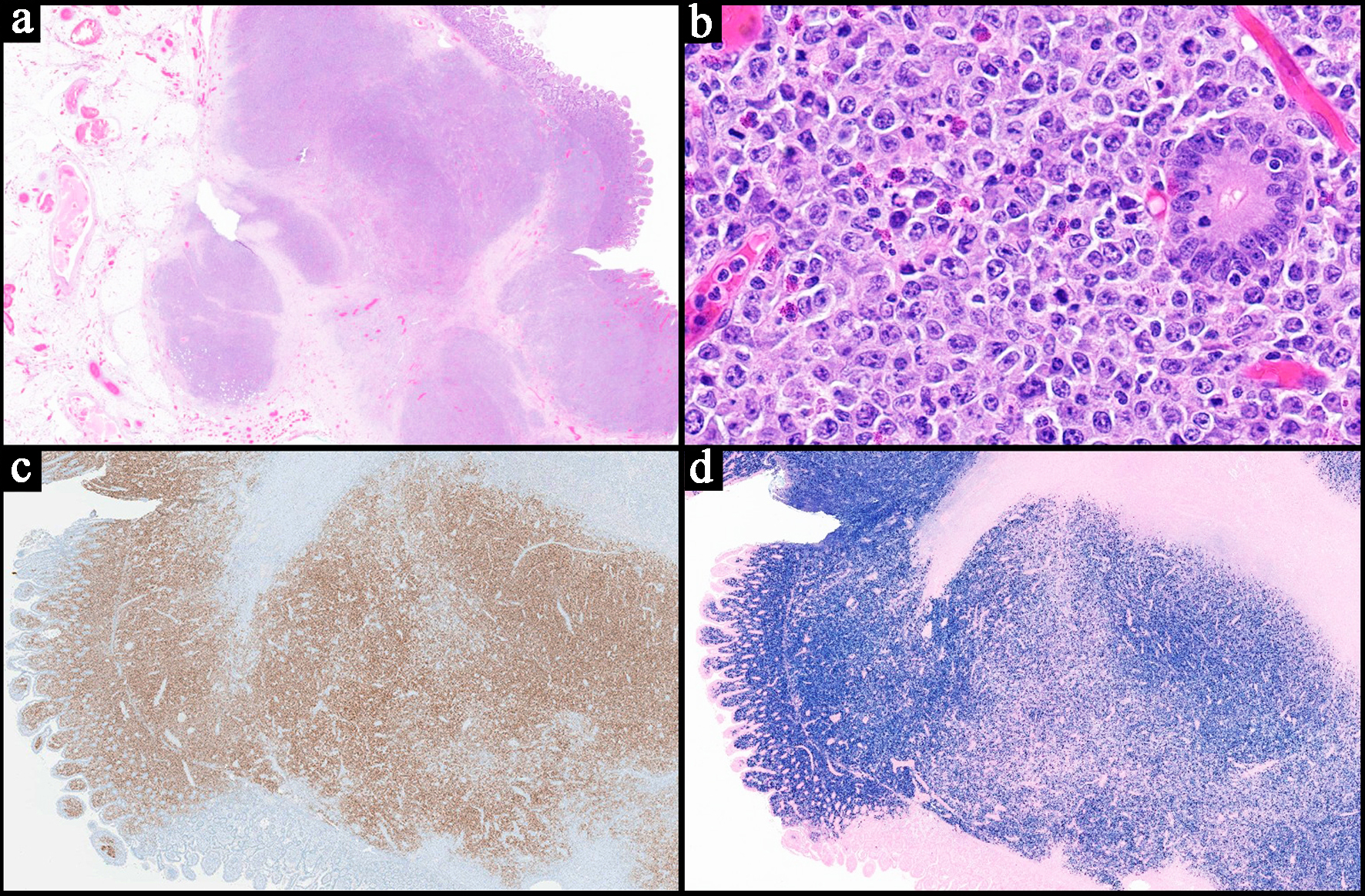 Click for large image |
Figure 7. Representative case of an EBV-positive diffuse large B-cell lymphoma subtype of the monomorphic post-transplant lymphoproliferative disorder. The patient had a history of solid organ transplant. (a) Low power view of a small bowel resection showing a transmural, diffuse, and partially nodular proliferation of neoplastic lymphoid cells (H&E, × 0.5). (b) High power view demonstrating large neoplastic cells with predominantly centroblastic morphology seen infiltrating around the intestinal glands. Mitotic figures and apoptotic bodies are present (H&E, × 40). (c) CD20 stain highlights the neoplastic cells, confirming B-cell lineage (× 1). (d) Epstein-Barr virus encoded RNA (EBER) in situ hybridization is positive in the majority of the neoplastic cells (approximately 90%) (× 1). H&E: hematoxylin and eosin. |
With regard to additional ancillary studies, molecular studies and cytogenetics may be of value for confirming the diagnosis. M-PTLD is commonly positive for BCL6 mutations and shows a complex karyotype [2]. BCL6 mutation can occur in some of the P-PTLDs [2].
Prognosis/treatment
The prognosis of PTLD is usually unfavorable with survival estimated between 30% and 60% [2]. Most patients present with advanced disease (Ann Arbor stage 3-4) [2, 48]. Prognosis depends on the extent of disease, location of disease, and patient variables [48]. Localized disease in the stomach appears to infer better prognosis [48]. Early PTLD development tends to have better prognosis than late [1]. Advances in management have recently improved outcomes [2]. Some cases can regress with reduction of immunosuppression, particularly those with EBV infection [2, 48, 51, 54]. Maintenance therapy includes tacrolimus, mycophenolate mofetil, and steroids (low dose) [51]. EBV-specific T-cell immunity or donor lymphocyte infusions have been utilized as second-line treatments for EBV-positive cases if immunosuppression reduction and chemotherapeutic options, such as rituximab, fail [48].
| Other B-Cell Lymphomas Involving the GI Tract | ▴Top |
While there are reports of other B-cell lymphomas involving the GI tract, the majority are usually secondary and in the context of systemic lymphoma. Although rare, several case reports have highlighted CLL/SLL involving the GI tract, but they were in the context of a systemic involvement by CLL/SLL [7, 55-57]. CLL/SLLs can present in various areas of the GI tract but appear to be mostly reported in the colon and usually show mucosal and submucosal lymphocytic infiltrates [55-57]. Rare reports have documented lymphoplasmacytic lymphoma/Waldenstrom’s macroglobulinemia involving the GI tract, but apart from single case reports, no larger studies have been conducted to investigate any unique trends/characteristics associated with these cases, likely because of their rarity [27, 58]. Two reports of ALK-positive large B-cell lymphoma have been reported, primarily involving the upper GI tract (stomach and duodenum) [59, 60]. These occurred in younger patients and showed infiltrative masses with neoplastic cells exhibiting immunoblastic and PB morphology [2, 59, 60]. The morphology can overlap with PB lymphoma and ALK immunostain can be helpful in distinguishing both [2]. Finally, rare reports have reported primary GI classic Hodgkin lymphoma with predilection of the upper GI tract [61, 62]. The morphology and immunophenotype match that of systemic Hodgkin lymphoma [62].
| Summarized Clinical and Diagnostic Approach to Primary GI Lymphomas | ▴Top |
The typical diagnostic approach for a primary GI B-cell lymphoma is best performed through correlation with a patient’s history, clinical presentation, imaging, endoscopic findings, and morphological assessment of tissue biopsy with associated ancillary studies. Although the findings in each entity can be variable, certain common features can help to guide the differential diagnosis. A summary of some of the key findings are outlined in Table 1. Higher grade diseases, such as Burkitt lymphoma and DLBCL, should be considered for patients presenting with large, rapidly growing masses with symptoms of abdominal pain, obstruction, and/or perforation. In contrast, MCL, MALT lymphoma, and FL typically have variable, non-specific findings or may even be asymptomatic and identified incidentally. A review of a patient’s history may reveal certain important etiological factors, such as previous solid organ/HSCT in PTLD, H. pylori infections in gastric MALT lymphoma and DLBCL, Campylobacter jejuni infections in small intestinal MALT lymphoma, and EBV infections in DLBCL, Burkitt lymphoma, PB lymphoma, and PTLD. Identification of a lesion through imaging and/or endoscopy in the stomach may raise the possibility of a MALT lymphoma or DLBCL, whereas FL is particularly common in the duodenum. Origin from the ileocecal region is common for Burkitt lymphoma. Endoscopy may show polypoid lesions with opaque white spots, enlarged villi, and dilated vessels in FL, whereas more aggressive entities may show large, ulcerated masses. Once the lesion has been identified, pathologic review of a tissue biopsy/excision is required for definitive diagnosis. An initial assessment of morphology may show characteristic features of nodular growth in FL, MCL, and MALT lymphoma. A mixture of centrocytes and centroblasts with positive expression of germinal center markers (e.g., CD10, BCL6) and BCL2 via immunohistochemistry (IHC) is characteristic of FL. In contrast, MCL is typically positive for cyclin D1, CD5, and SOX11, whereas MALT lymphoma is classically negative for CD5 and CD10. More aggressive lymphomas like DLBCL, Burkitt lymphoma, and PB lymphoma commonly show diffuse sheets of neoplastic cells with high proliferation rates. In these scenarios, plasma cell markers (e.g., CD38, CD138) can help identify PB lymphoma. Germinal center marker expression, strong MYC, and absence of BCL2 are characteristic of Burkitt lymphoma, and these markers should also be included in the investigation of lymphomas that show higher grade morphology.
| Conclusion | ▴Top |
In summary, this review offers an overview of the common primary GI B-cell lymphomas, discussing their clinicopathological features and highlighting some of their unique characteristics. As ongoing research efforts continue, we suspect that the list of primary GI B-cell lymphomas will expand, and our understanding of their biology, clinicopathological manifestations, and therapeutic approaches will continue to evolve.
Acknowledgments
None to declare.
Financial Disclosure
This project did not receive any funding.
Conflict of Interest
None to declare.
Author Contributions
Conception and design: IE. Drafting of article: IE and CL. Editing/formatting: IE. Creating figures: IE and CL. Creating table: CL. Reviewing and editing: IE and CL. Supervision: IE. Approval of final manuscript: IE and CL.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Hanafy AK, Morani AC, Menias CO, Pickhardt PJ, Shaaban AM, Mujtaba B,
Mellnick VM, et al. Hematologic malignancies of the gastrointestinal luminal tract. Abdom Radiol
(NY). 2020;45(10):3007-3027.
doi pubmed - Fukayama M, Chan John KC. Haematolymphoid tumours of the digestive system. WHO Classification of Tumours Editorial Board. Digestive system tumours. Lyon (France): International Agency for Research on Cancer; 2019. WHO classification of tumours series, 5th ed.; vol. 1. Available from: https://tumourclassification.iarc.who.int/chapters/31.
- Shen XZ, Liu F, Ni RJ, Wang BY. Primary histiocytic sarcoma of the
stomach: a case report with imaging findings. World J Gastroenterol.
2013;19(3):422-425.
doi pubmed - Elsharawi I, Williams S, Stueck A. Non-mass-forming myeloid sarcoma
of the colon presenting with diarrhea. ACG Case Rep J. 2023;10(12):e01213.
doi pubmed - Vannata B, Zucca E. Primary extranodal B-cell lymphoma: current
concepts and treatment strategies. Chin Clin Oncol. 2015;4(1):10.
doi pubmed - Yang H, Xun Y, Ke C, Tateishi K, You H. Extranodal lymphoma:
pathogenesis, diagnosis and treatment. Mol Biomed. 2023;4(1):29.
doi pubmed - Ghimire P, Wu GY, Zhu L. Primary gastrointestinal lymphoma.
World J Gastroenterol. 2011;17(6):697-707.
doi pubmed - Dawson IM, Cornes JS, Morson BC. Primary malignant lymphoid tumours
of the intestinal tract. Report of 37 cases with a study of factors influencing prognosis.
Br J Surg. 1961;49:80-89.
doi pubmed - Rohatiner A, d'Amore F, Coiffier B, Crowther D, Gospodarowicz M,
Isaacson P, Lister TA, et al. Report on a workshop convened to discuss the pathological and
staging classifications of gastrointestinal tract lymphoma. Ann Oncol.
1994;5(5):397-400.
doi pubmed - Iwamuro M, Kondo E, Takata K, Yoshino T, Okada H. Diagnosis of
follicular lymphoma of the gastrointestinal tract: A better initial diagnostic workup.
World J Gastroenterol. 2016;22(4):1674-1683.
doi pubmed - Hayashi H, Onishi Y, Mitsuoka H, Ogura T, Maeda M, Nishigami T,
Harada M. Regression of follicular lymphoma of the duodenum following eradication of H. pylori
infection. Intern Med. 2013;52(23):2611-2614.
doi pubmed - Toyoda H, Yamaguchi M, Nakamura S, Nakamura T, Kimura M, Suzuki H,
Mukai K, et al. Regression of primary lymphoma of the ampulla of Vater after eradication of
Helicobacter pylori. Gastrointest Endosc. 2001;54(1):92-96.
doi pubmed - Nakase H, Matsuura M, Mikami S, Chiba T. Magnified endoscopic view of
primary follicular lymphoma at the duodenal papilla. Intern Med. 2007;46(3):141-142.
doi pubmed - Matysiak-Budnik T, Jamet P, Chapelle N, Fabiani B, Coppo P,
Ruskone-Fourmestraux A. Primary gastrointestinal follicular lymphomas: a prospective study of 31
patients with long-term follow-up registered in the French Gastrointestinal Lymphoma Study Group
(GELD) of the French Federation of Digestive Oncology (FFCD). Gut Liver.
2022;16(2):207-215.
doi pubmed - Iwamuro M, Tanaka T, Ennishi D, Otsuka M. Recent updates on treatment
options for primary follicular lymphoma of the gastrointestinal tract. Expert Rev Gastroenterol
Hepatol. 2024;18(7):367-375.
doi pubmed - Mohammed A, Shariati F, Paranji N, Waghray N. Primary follicular
lymphoma of colon: A case series and review of literature. Clin Case Rep.
2021;9(7):e04486.
doi pubmed - Castellino A, Tun AM, Wang Y, Habermann TM, King RL, Ristow KM,
Cerhan JR, et al. Clinical characteristics and outcomes of primary versus secondary
gastrointestinal mantle cell lymphoma. Blood Cancer J. 2021;11(1):8.
doi pubmed - Dasappa L, Suresh Babu MC, Sirsath NT, Suresh TM, Govind Babu K,
Sathyanarayna V, Lokesh KN, et al. Primary gastrointestinal mantle cell lymphoma: a
retrospective study. J Gastrointest Cancer. 2014;45(4):481-486.
doi pubmed - Petranovic D, Pilcic G, Peitl M, Cubranic A, Valkovic T, Nacinovic
AD, Lucin K, et al. Primary gastric mantle cell lymphoma. Hematol Rep. 2012;4(1):e1.
doi pubmed - Elsharawi I, Selegean S, Carter M. Utility of p53 immunohistochemical
staining for risk stratification of mantle cell lymphoma. J Hematol.
2024;13(5):200-206.
doi pubmed - Small S, Barnea Slonim L, Williams C, Karmali R. B Cell Lymphomas of
the GI Tract. Curr Gastroenterol Rep. 2021;23(7):9.
doi pubmed - Zucca E, Bertoni F. The spectrum of MALT lymphoma at different sites:
biological and therapeutic relevance. Blood. 2016;127(17):2082-2092.
doi pubmed - Ishikawa E, Nakamura M, Satou A, Shimada K, Nakamura S.
Mucosa-Associated Lymphoid Tissue (MALT) lymphoma in the gastrointestinal tract in the modern
era. Cancers (Basel). 2022;14(2):446.
doi pubmed - Nakamura S, Hojo M. Diagnosis and treatment for gastric
mucosa-associated lymphoid tissue (MALT) lymphoma. J Clin Med. 2022;12(1):120.
doi pubmed - Nagesh VK, Bhuju R, Mohammed AS, Martinez E, Basta M, Francis D, Dey
S, et al. Gastrointestinal lymphomas: a comprehensive review of epidemiology, clinical features,
diagnosis, histopathology, and management. Lymphatics. 2025;3(4):31.
doi - Bahceci D, Wen KW. Extranodal marginal zone lymphoma of mucosa associated lymphoid tissue (MALT lymphoma). PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/stomachmalt.html.
- Attallah HS, Moonim M, Fields P, Wrench D, Brady J, Mikhaeel NG.
Primary isolated lymphoplasmacytic lymphoma (LPL) of the stomach: a case report.
Am J Case Rep. 2020;21:e921840.
doi pubmed - Bai Z, Zhou Y. A systematic review of primary gastric diffuse large
B-cell lymphoma: Clinical diagnosis, staging, treatment and prognostic factors. Leuk Res.
2021;111:106716.
doi pubmed - Chen X, Wang J, Liu Y, Lin S, Shen J, Yin Y, Wang Y. Primary
intestinal diffuse large B-cell lymphoma: novel insights and clinical perception. Front Oncol.
2024;14:1404298.
doi pubmed - Suresh B, Asati V, Lakshmaiah KC, Babu G, Lokanatha D, Jacob LA,
Lokesh KN, et al. Primary gastrointestinal diffuse large B-cell lymphoma: a prospective study
from South India. South Asian J Cancer. 2019;8(1):57-59.
doi pubmed - Lewis CS, Joy G, Jensen P, Barraclough A, Franco N, Talaulikar D,
Hawkes EA, et al. Primary gastric diffuse large B-cell lymphoma: A multicentre retrospective
study. Br J Haematol. 2024;205(2):534-541.
doi pubmed - Yang H, Wu M, Shen Y, Lei T, Mi L, Leng X, Ping L, et al. Treatment
strategies and prognostic factors of primary gastric diffuse large B cell lymphoma: a
retrospective multicenter study of 272 cases from the China lymphoma patient registry.
Int J Med Sci. 2019;16(7):1023-1031.
doi pubmed - Iwamuro M, Tanaka T, Okada H. Review of lymphoma in the duodenum: an
update of diagnosis and management. World J Gastroenterol.
2023;29(12):1852-1862.
doi pubmed - Chen SY, Xu PP, Feng R, Cui GH, Wang L, Cheng S, Mu RJ, et al.
Extranodal diffuse large B-cell lymphoma: Clinical and molecular insights with survival outcomes
from the multicenter EXPECT study. Cancer Commun (Lond). 2025;45(8):919-935.
doi pubmed - Rumney S, Rajesh A, Brigmon E. Primary gastrointestinal diffuse large
B-cell lymphoma presenting as ileal perforation. Cureus. 2023;15(4):e37341.
doi pubmed - Maeshima AM, Taniguchi H, Ito Y, Hatta S, Suzuki T, Yuda S, Makita S,
et al. Clinicopathological characteristics of diffuse large B-cell lymphoma involving small and
large intestines: an analysis of 126 patients. Int J Hematol.
2019;110(3):340-346.
doi pubmed - Owattanapanich W, Ruchutrakool T, Pongpruttipan T, Maneerattanaporn
M. A 10-year cohort study of 175 primary gastrointestinal lymphoma cases in Thailand: clinical
features and outcomes in the immunochemotherapy era. Hematology. 2021;26(1):249-255.
doi pubmed - Xie Y, Jia M, Shi J, Tao Y. Inferior prognosis of gastric involvement
in patients with gastrointestinal Burkitt Lymphoma. Cancer Med. 2020;9(9):3107-3114.
doi pubmed - Rodrigues-Fernandes CI, Perez-de-Oliveira ME, Aristizabal Arboleda
LP, Fonseca FP, Lopes MA, Vargas PA, Santos-Silva AR. Clinicopathological analysis of oral
Burkitt's lymphoma in pediatric patients: A systematic review. Int J Pediatr
Otorhinolaryngol. 2020;134:110033.
doi pubmed - Zhang Z, Yan J, Yan J, Huang L, Chen Y, Ni X. Surgical management of
children with Burkitt's lymphoma involving the gastrointestinal tract. Pediatr Surg Int.
2024;41(1):44.
doi pubmed - Kapitancuke M, Vasciunaite A, Augustiniene R, Sakalinskiene J,
Kleinotiene G. Primary gastric Burkitt lymphoma-induced anaemia: a case report and a literature
review. Acta Med Litu. 2018;25(1):23-30.
doi pubmed - Khan A, Khan S, Arshad U. Primary gastric Burkitt's LYMPHOMA.
Pak J Med Sci. 2017;33(5):1294-1297.
doi pubmed - Cubranic A, Golcic M, Fuckar-Cupic D, Brozovic B, Gajski D, Brumini
I. Burkitt lymphoma in gastrointestinal tract: a report of two cases. Acta Clin Croat.
2019;58(2):386-390.
doi pubmed - Luria L, Nguyen J, Zhou J, Jaglal M, Sokol L, Messina JL, Coppola D,
et al. Manifestations of gastrointestinal plasmablastic lymphoma: a case series with literature
review. World J Gastroenterol. 2014;20(33):11894-11903.
doi pubmed - Fu Z, Wang H, Lauwers GY, Jiang K, Jayaratne NL, Bridglal S, Dong N,
et al. Primary plasmablastic lymphoma of the gastrointestinal tract: A series of 13 HIV-negative
cases and a review of literature. Ann Diagn Pathol. 2023;67:152204.
doi pubmed - Gianarakis M, Rizvi SS, Raza HS, Ghoulam E, Carroll RE. S3143
Gastrointestinal manifestations of plasmablastic lymphoma: a case series from a tertiary cancer
center. The American Journal of Gastroenterology. 2024;119(10S):S2129-S2130.
doi - Castillo JJ, Bibas M, Miranda RN. The biology and treatment of
plasmablastic lymphoma. Blood. 2015;125(15):2323-2330.
doi pubmed - Reiche W, Tauseef A, Sabri A, Mirza M, Cantu D, Silberstein P,
Chandan S. Gastrointestinal manifestations, risk factors, and management in patients with
post-transplant lymphoproliferative disorder: A systematic review.
World J Transplant. 2022;12(8):268-280.
doi pubmed - Hsu YC, Liao WC, Wang HP, Yao M, Lin JT. Catastrophic
gastrointestinal manifestations of post-transplant lymphoproliferative disorder. Dig Liver Dis.
2009;41(3):238-241.
doi pubmed - Curtis RE, Travis LB, Rowlings PA, Socie G, Kingma DW, Banks PM,
Jaffe ES, et al. Risk of lymphoproliferative disorders after bone marrow transplantation: a
multi-institutional study. Blood. 1999;94(7):2208-2216.
pubmed - Wozniak LJ, Mauer TL, Venick RS, Said JW, Kao RL, Kempert P, Marcus
EA, et al. Clinical characteristics and outcomes of PTLD following intestinal transplantation.
Clin Transplant. 2018;32(8):e13313.
doi pubmed - Khedmat H, Amini M, Ghamar-Chehreh ME. Colorectal involvement by
post-transplant lymphoproliferative disorders: a review of 81 cases.
Saudi J Kidney Dis Transpl. 2014;25(3):597-604.
doi pubmed - Morscio J, Tousseyn T. Recent insights in the pathogenesis of
post-transplantation lymphoproliferative disorders. World J Transplant.
2016;6(3):505-516.
doi pubmed - Cassidy D, Chapman J. PTLD-polymorphic. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/lymphomanonbpolymorphicb.html.
- Arkkila PE, Nuutinen H, Ebeling F, Elonen E, Karkkainen P,
Karjalainen-Lindsberg ML. Colonic involvement in a patient with chronic lymphocytic leukaemia.
Gastroenterol Res Pract. 2008;2008:742146.
doi pubmed - Abella E, Gimenez T, Gimeno J, Cervera M, Pedro C, Gimeno E, Alvarez
A, et al. Diarrheic syndrome as a clinical sign of intestinal infiltration in progressive B-cell
chronic lymphocytic leukemia. Leuk Res. 2009;33(1):159-161.
doi pubmed - Cabuk D, Balli F, Yilmaz Y, Duman AE, Uygun K. Gastrointestinal
involvement in a patient with chronic lymphocytic leukemia. Balkan Med J.
2019;37(1):50-51.
doi pubmed - Bedine MS, Yardley JH, Elliott HL, Banwell JG, Hendrix TR. Intestinal
involvement in Waldenstrom's macroglobulinemia. Gastroenterology. 1973;65(2):308-315.
pubmed - McManus DT, Catherwood MA, Carey PD, Cuthbert RJ, Alexander HD.
ALK-positive diffuse large B-cell lymphoma of the stomach associated with a clathrin-ALK
rearrangement. Hum Pathol. 2004;35(10):1285-1288.
doi pubmed - Xing X, Lin D, Ran W, Liu H. ALK-positive diffuse large B-cell
lymphoma of the duodenum: A case report and review of the literature. Exp Ther Med.
2014;8(2):409-412.
doi pubmed - Sharma S, Rana S, Kapur S, Jairajpuri ZS. Primary intestinal
Hodgkin's lymphoma: an uncommon presentation. J Lab Physicians.
2013;5(2):124-126.
doi pubmed - Kilaru S, Panda SS, Das H, Sahoo N, Mohapatra D, Mohapatra SSG,
Kolluri S. Primary gastric Hodgkin's lymphoma: a rare coincidence. Cancer Treat Res Commun.
2020;24:100194.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Hematology is published by Elmer Press Inc.