| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://jh.elmerpub.com |
Review
Volume 14, Number 5, October 2025, pages 253-256

Iron-Induced Hypophosphatemia: A Review of Pathophysiology, Drug Safety, and Pharmacogenomic Perspectives
Simone Pozzesserea, b
aUOC Terapie Cellulari e Officina Trasfusionale, Dipartimento Terapie Cellulari,
Ematologia e Medicina di Laboratorio, Azienda Ospedaliera Universitaria Senese,
Italy
bUnita Operativa Complessa di Immunoematologia, Medicina Trasfusionale e
Laboratorio, Azienda Ospedaliera Universitaria Meyer, Firenze, Italy
Manuscript submitted July 19, 2025, accepted September 1, 2025, published online October 10,
2025
Short title: Iron-Induced Hypophosphatemia
doi: https://doi.org/10.14740/jh2111
| Abstract | ▴Top |
Intravenous (IV) iron therapy is a cornerstone in the management of iron deficiency anemia, yet its administration is associated with significant side effects, most notably hypophosphatemia. This adverse event is not a class effect but is disproportionately linked to specific formulations, particularly ferric carboxymaltose (FCM). This review synthesizes the current clinical evidence, elucidates the central pathophysiological role of fibroblast growth factor 23 (FGF23), and explores a potential pharmacogenomic basis for individual susceptibility. Clinical data from numerous randomized controlled trials and meta-analyses confirm that FCM induces hypophosphatemia in over 50% of patients, a rate far exceeding that of other IV iron preparations. The proposed underlying mechanism involves a “two-hit” process: pre-existing iron deficiency upregulates FGF23 gene transcription, while FCM administration is thought to inhibit the proteolytic cleavage of the FGF23 protein. This uncoupling of production and degradation leads to a surge in active, intact FGF23 (iFGF23), causing renal phosphate wasting. We explore the potential for genetic polymorphisms in FGF23 and its key processing enzymes, such as FURIN, GALNT3, and FAM20C, to modulate individual risk. Understanding this complex interplay is crucial for risk stratification, appropriate formulation selection, and patient monitoring to mitigate the acute and chronic consequences of iatrogenic hypophosphatemia, including debilitating fatigue and osteomalacia.
Keywords: Ferric carboxymaltose; Hypophosphatemia; FGF23; Drug safety; Pharmacogenomics; Adverse drug reaction; Iron deficiency anemia; Hematology
| Introduction | ▴Top |
Iron deficiency anemia (IDA) is a global health issue, and intravenous (IV) iron preparations are essential when oral therapy is ineffective or not tolerated [1]. While modern formulations have improved safety profiles, they are not without adverse effects. A significant and increasingly recognized complication is hypophosphatemia, a condition characterized by low serum phosphate levels [2]. This adverse event can range from an asymptomatic laboratory finding to a severe, debilitating condition causing fatigue, muscle weakness, and, with chronic exposure, osteomalacia and fractures [3].
Crucially, the risk of hypophosphatemia is not uniform across all IV iron products. Evidence clearly indicates that ferric carboxymaltose (FCM) and iron polymaltose are associated with a significantly higher incidence and severity of hypophosphatemia compared to other formulations like ferric derisomaltose (FDI) or iron sucrose [4, 5]. This suggests a mechanism tied to the specific properties of certain iron-carbohydrate complexes rather than a class effect of iron repletion itself [2].
The pathophysiology points to a complex hormonal dysregulation centered on fibroblast growth factor 23 (FGF23), a key regulator of phosphate homeostasis [6]. This review aims to: 1) systematically analyze the clinical evidence comparing different IV iron formulations; 2) detail the proposed pathophysiological mechanism involving FGF23 dysregulation; 3) explore a potential pharmacogenomic basis for individual susceptibility based on an analysis of relevant genes; and 4) discuss the clinical implications for drug safety and patient management in hematology practice.
| Clinical Evidence and Formulation-Specific Risk | ▴Top |
The link between IV iron and hypophosphatemia is now firmly established through multiple randomized controlled trials (RCTs) and meta-analyses. These studies consistently demonstrate that FCM carries the highest risk [7]. A landmark RCT comparing FCM to ferumoxytol (FMX) found that 50.8% of patients receiving FCM developed hypophosphatemia (< 2.0 mg/dL), compared to just 0.9% in the FMX group [8]. Similarly, the PHOSPHARE-IBD trial, which compared FCM to FDI, reported hypophosphatemia incidence rates of 51.0% for FCM versus 8.3% for FDI [9]. A network meta-analysis confirmed FCM as the formulation with the highest risk, ranking it significantly worse than FDI, iron sucrose, and FMX [7]. It should be noted that iron polymaltose, another formulation, has also been associated with a high risk of FGF23-mediated hypophosphatemia [5, 10].
The onset of hypophosphatemia typically occurs around 2 weeks post-infusion, which corresponds to the peak effect of FGF23 on renal tubules [10]. While often transient, the duration is variable. Many cases resolve within 5 to 12 weeks, but a significant proportion of patients (up to 45%) can experience persistent hypophosphatemia that lasts for months, particularly after repeated infusions [8, 11].
Several clinical factors influence the risk of developing this complication. While treatment with a high-risk formulation like FCM is the primary determinant for incident hypophosphatemia, other factors may modulate the risk of developing severe or persistent forms [12]. Paradoxically, normal renal function (a higher estimated glomerular filtration rate (eGFR)) is a major risk factor for the initial event, as functioning kidneys are required to excrete phosphate under the influence of FGF23 [6]. Other identified risks include more severe baseline iron deficiency and the need for repeated doses of FCM, which can lead to a cumulative effect [2]. The evidence for pre-existing vitamin D deficiency as a major risk factor is limited and largely based on case reports and reviews [3]. The symptoms of hypophosphatemia (e.g., fatigue, weakness) can also be mistaken for those of the underlying anemia, leading to diagnostic confusion and potentially inappropriate re-treatment [9].
| Pathophysiology: The Proposed Central Role of FGF23 | ▴Top |
The pathophysiology of this adverse event is not due to direct iron toxicity but is best explained by a plausible “two-hit hypothesis” involving FGF23 [2]. It is important to note that while this model fits the clinical data, direct molecular evidence of specific iron formulations inhibiting the cleavage machinery is still lacking.
First hit: iron deficiency
Iron deficiency itself stimulates the transcription of the FGF23 gene in osteocytes, likely mediated by hypoxia-inducible factors (HIFs) [13]. However, this does not cause hypophosphatemia because the increased production of the FGF23 protein is matched by a compensatory increase in its proteolytic cleavage [14]. This cleavage splits the active, intact FGF23 (iFGF23) into inactive fragments. Thus, in iron deficiency, levels of C-terminal FGF23 (cFGF23) are high, but iFGF23 levels remain normal, preserving phosphate balance [13].
Second hit: FCM administration
The administration of FCM is thought to deliver the second hit by inhibiting the proteolytic cleavage of FGF23 [15]. The unique carbohydrate shell of FCM is hypothesized to be responsible for this effect, possibly by sterically hindering the enzymes that cleave the protein [4]. This blocks the compensatory degradation pathway, leading to a rapid and sustained surge in circulating levels of active iFGF23 [6]. This excess iFGF23 then acts on the kidneys to suppress the NaPi-IIa and NaPi-IIc phosphate co-transporters, causing massive renal phosphate wasting (phosphaturia) and a sharp drop in serum phosphate [14].
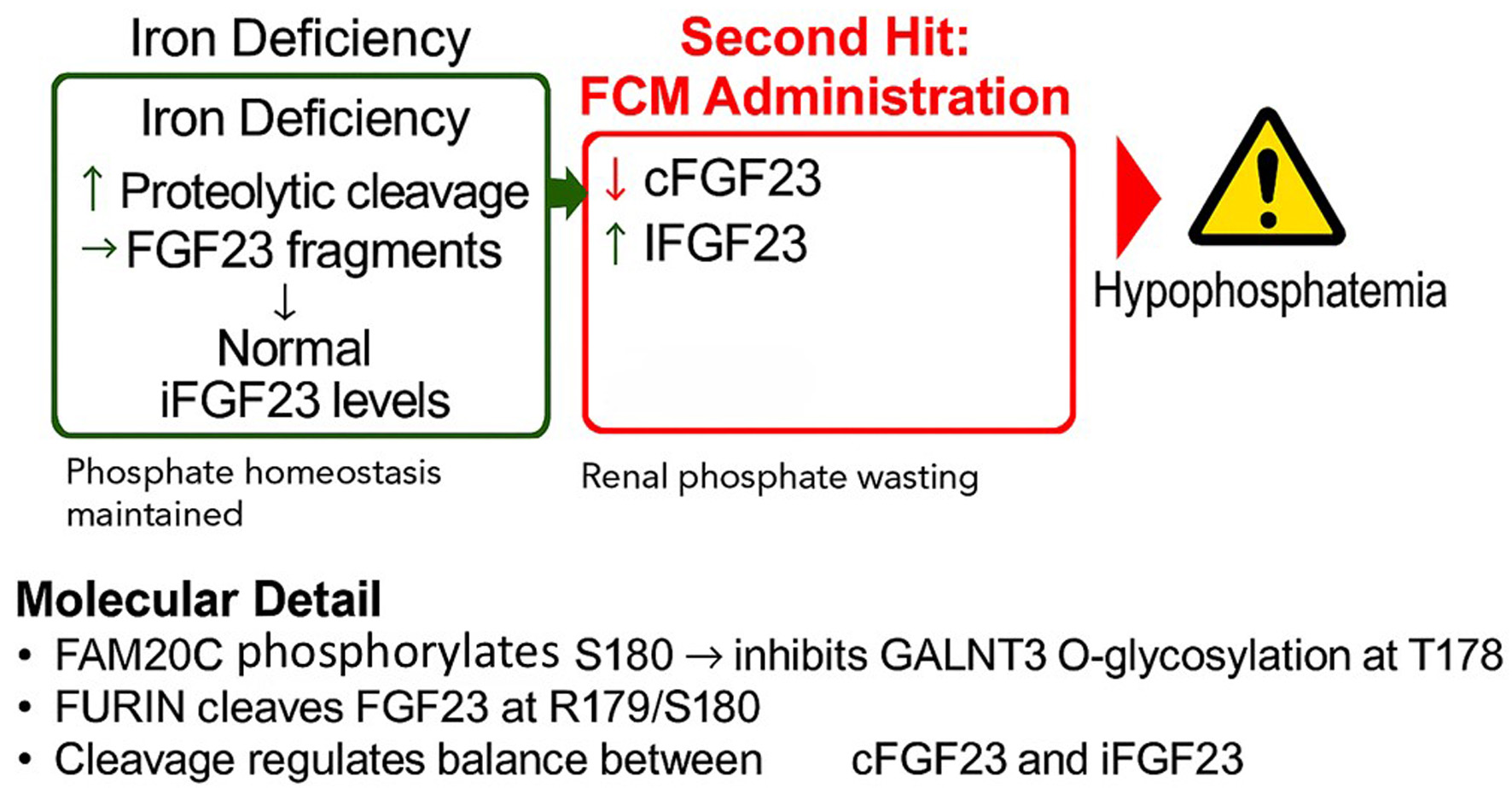
Figure 1 shows a schematic representation of this mechanism.
 Click for large image |
Figure 1. The proposed “two-hit” mechanism of ferric carboxymaltose (FCM)-induced hypophosphatemia. (a) In a state of iron deficiency, transcription of the FGF23 gene is upregulated in osteocytes, leading to increased protein production. However, this is matched by a compensatory increase in proteolytic cleavage, resulting in high levels of inactive C-terminal FGF23 (cFGF23) fragments but normal levels of active, intact FGF23 (iFGF23). Phosphate homeostasis is maintained. (b) The administration of FCM is hypothesized to inhibit the cleavage process. This uncouples the high production rate from degradation, leading to a surge in circulating iFGF23, which in turn causes renal phosphate wasting and hypophosphatemia. (c) Molecular detail of the FGF23 cleavage site. The protease FURIN cleaves the protein at R179/S180. This cleavage is promoted by the kinase FAM20C, which phosphorylates serine 180 (S180). This phosphorylation prevents the protective O-glycosylation of threonine 178 (T178) by the enzyme GALNT3. |
| A Potential Pharmacogenomic Basis for Susceptibility | ▴Top |
While the choice of iron formulation is the primary risk factor, the high incidence but variable severity of FCM-induced hypophosphatemia suggests a potential role for genetic predisposition. This section explores this as a speculative but biologically plausible area for future research.
The post-translational processing of FGF23 is a critical control point. The cleavage of FGF23 occurs at a specific site (R176HTR179↓S180) and is regulated by a complex interplay of enzymes. This process is performed primarily by the enzyme furin [16]. However, its activity is modulated by two other key proteins: the glycosyltransferase GALNT3, which protects the protein from being cleaved by adding a sugar moiety to threonine-178 [17], and the kinase FAM20C, which phosphorylates serine 180. This phosphorylation promotes cleavage by preventing the protective action of GALNT3 [18]. Genetic variants in the genes encoding these four proteins - FGF23, FURIN, GALNT3, and FAM20C - could therefore logically alter an individual’s susceptibility.
Known rare diseases provide a biological rationale that inferentially suggests a possible mechanism for genetic predisposition. Autosomal dominant hypophosphatemic rickets (ADHR) is caused by mutations in the FGF23 cleavage site that make the protein resistant to furin, leading to chronic iFGF23 excess [19]. Conversely, familial tumoral calcinosis (HFTC) can be caused by loss-of-function mutations in GALNT3 or FAM20C, leading to excessive FGF23 cleavage and hyperphosphatemia [17, 18].
We hypothesize that common, less impactful single nucleotide polymorphisms (SNPs) in these genes could create a “polygenic risk score”. An individual carrying a combination of variants that slightly alter the function of these enzymes may have a “fragile” processing system. While stable under normal conditions, this system could be more easily overwhelmed by the cleavage-inhibiting effect of FCM. Future pharmacogenomic studies would be needed to validate these candidate variants and explore this hypothesis [20].
| Conclusion and Clinical Implications | ▴Top |
FCM-induced hypophosphatemia is a frequent and clinically significant adverse drug reaction that remains underappreciated in routine hematology practice. The evidence strongly indicates that this is not a class effect of IV iron but a specific liability of certain formulations, driven by a proposed interference with FGF23 cleavage. The most insidious aspect of this adverse event is its ability to create a diagnostic quagmire, where symptoms of hypophosphatemia are mistaken for the underlying anemia, potentially leading to re-administration of the causative agent.
The current standard of care should evolve to reflect this evidence. A proactive strategy is essential. The most effective and practical strategy for primary prevention is the selection of an IV iron formulation with a proven low risk of hypophosphatemia, such as FDI or iron sucrose. When high-risk formulations like FCM are used, clinicians must be educated to recognize the high-risk patient profile and consider monitoring serum phosphate, especially before administering subsequent doses.
Looking forward, a potential future direction for research lies in pharmacogenomics. While our hypothesis of a polygenic risk model is biologically plausible, it remains speculative. It is important to acknowledge that the implementation of such genetic testing faces significant practical hurdles, including cost and reimbursement, and its clinical utility is not yet clear. Therefore, framing this as an immediate “next frontier” would be premature. Nevertheless, understanding the potential genetic basis of this adverse reaction remains an important gap in the literature that future studies, incorporating DNA collection, could help to address. A personalized approach, combining clinical risk stratification with, perhaps one day, genetic insights, would be the ultimate path to maximizing the benefit and minimizing the risk of this essential therapy.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Author Contributions
SP is the sole author and was responsible for all aspects of the manuscript’s conception, research, writing, and final approval.
Data Availability
The author declares that data supporting the findings of this study are available within the article.
Generative Artificial Intelligence (AI) Disclosure
In compliance with the journal’s policies, the author(s) disclose the use of a large language model (Google) during the preparation of this manuscript. The AI tool was utilized for the purposes of reviewing the manuscript for clarity, structure, and language, as well as for assistance with formatting the document according to the journal’s specific guidelines. The author(s) take full responsibility for the final content and all scientific conclusions presented.
| References | ▴Top |
- Auerbach M, DeLoughery T. Management of iron deficiency. Hematology Am Soc Hematol Educ Program. 2019;2019(1):315-322.
- Pollock E, Kalra PA. The pathophysiology of intravenous iron-induced hypophosphataemia. J Ren Care. 2021;47(1):4-10.
- Hardy S, Vandemergel X. Intravenous iron-induced hypophosphatemia: a review of the literature. Expert Opin Drug Saf. 2021;20(9):1-12.
- Blazevic A, Hunze J, Schlosser M, et al. Ferric carboxymaltose-induced hypophosphatemia: a review of the evidence, pathophysiology, and clinical implications. Am J Ther. 2021;28(4):e477-e486.
- Schouten BJ, Hunt PJ, Livesey JH, Frampton CM, Soule SG. FGF23
elevation and hypophosphatemia after intravenous iron polymaltose: a prospective study.
J Clin Endocrinol Metab. 2009;94(7):2332-2337.
doi pubmed - Martens KL, Wolf M. Incidence, mechanism, and consequences of IV
iron-induced hypophosphatemia. Hematology Am Soc Hematol Educ Program.
2023;2023(1):636-639.
doi pubmed - Bellos I, Frountzas M, Pergialiotis V. Comparative Risk of
Hypophosphatemia Following the Administration of Intravenous Iron Formulations: A Network
Meta-Analysis. Transfus Med Rev. 2020;34(3):188-194.
doi pubmed - Wolf M, Chertow GM, Macdougall IC, Kaper R, Krop J, Strauss W.
Randomized trial of intravenous iron-induced hypophosphatemia. JCI Insight.
2018;3(23):e124486.
doi pubmed - Zoller H, Wolf M, Blumenstein I, Primas C, Lindgren S, Thomsen LL,
Reinisch W, et al. Hypophosphataemia following ferric derisomaltose and ferric carboxymaltose in
patients with iron deficiency anaemia due to inflammatory bowel disease (PHOSPHARE-IBD): a
randomised clinical trial. Gut. 2023;72(4):644-653.
doi pubmed - Schaefer B, Zoller H, Wolf M. Risk factors for and effects of
persistent and severe hypophosphatemia following ferric carboxymaltose. J Clin Endocrinol
Metab. 2022;107(4):1009-1019.
doi pubmed - Schaefer B, Tobiasch M, Wagner S, Glodny B, Tilg H, Wolf M, Zoller H.
Hypophosphatemia after intravenous iron therapy: Comprehensive review of clinical findings and
recommendations for management. Bone. 2022;154:116202.
doi pubmed - Clinkenbeard EL, Farrow EG, Summers LJ, et al. Iron deficiency and the role of the erythropoietin-FGF23-hepcidin axis in the development of anemia. Am J Physiol Renal Physiol. 2016;311(5):F927-F936.
- Martin A, David V, Quarles LD. Regulation and function of the
FGF23/klotho endocrine pathways. Physiol Rev. 2012;92(1):131-155.
doi pubmed - Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron
modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy
humans. J Clin Endocrinol Metab. 2011;96(11):3541-3549.
doi pubmed - Coppolino G, Comi A, Rivoli L, et al. Iron infusion and induced hypophosphatemia: the role of FGF23. J Renal Inj Prev. 2019;8(4):e01.
- Latic E, Erclik M, Thi Tran T, et al. Furin, a proprotein convertase, is only partially responsible for FGF23 cleavage. Front Endocrinol (Lausanne). 2021;12:690681.
- Kato K, Jeanneau C, Tarp MA, Benet-Pages A, Lorenz-Depiereux B,
Bennett EP, Mandel U, et al. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis.
Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem.
2006;281(27):18370-18377.
doi pubmed - Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda
Appaiah H, Koller A, et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3
glycosylation, and furin proteolysis. Proc Natl Acad Sci U S A.
2014;111(15):5520-5525.
doi pubmed - ADHR Consortium. Autosomal dominant hypophosphataemic rickets is
associated with mutations in FGF23. Nat Genet. 2000;26(3):345-348.
doi pubmed - Cacace E, Mignogna C, Peluso S, et al. Genetic background of adverse drug reactions. Front Pharmacol. 2021;12:651720.
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Hematology is published by Elmer Press Inc.