| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://jh.elmerpub.com |
Review
Volume 14, Number 4, August 2025, pages 174-192

Mast Cell Leukemia: Comprehensive Review of Literature With Current Insights and Updates on Management
Muralidhar Idamakantia, d ![]() , Ala Ebaidb, Rani Indrani
Bijjama, Alexei Bakhirevc
, Ala Ebaidb, Rani Indrani
Bijjama, Alexei Bakhirevc
aAdult Internal Medicine Services (AIMS), Presbyterian Healthcare Services (PHS),
Albuquerque, NM 87106, USA
bDivision of Hematology/Oncology, The University of
New Mexico Comprehensive Cancer Center (UNMCC), Albuquerque, NM 87102,
USA
cPathology Associates of Albuquerque (PAA), Albuquerque, NM 87106,
USA
dCorresponding Author: Muralidhar Idamakanti, Adult Internal Medicine
Services (AIMS), Presbyterian Healthcare Services (PHS), Albuquerque, NM 87106, USA
Manuscript submitted June 20, 2025, accepted August 8, 2025, published online August 25,
2025
Short title: Review of Mast Cell Leukemia
doi: https://doi.org/10.14740/jh2104
| Abstract | ▴Top |
Mast cell leukemia (MCL) is an exceedingly rare and aggressive variant of systemic mastocytosis (SM). MCL is classified as primary, occurring de novo without prior mast cell (MC) disorders or secondary, from a pre-existing SM, and acute aggressive form with C-findings that indicate organ damage or chronic indolent form without organ damage. Of the cases, 60-65% are aleukemic with < 10% circulating MCs in the peripheral blood, and the rest of the cases are leukemic with > 10% MCs. Diagnosis is typically confirmed by bone marrow biopsy revealing greater than 20% atypical or immature MCs in the smear. Specific MCL-targeted treatments are limited, and multiple treatment modalities used for SM and acute myeloid leukemia (AML) have been tried in MCL with limited success and variable survival benefit. The management has significantly advanced since the implication of the receptor tyrosine kinase type III (KIT) D816V mutation in the pathogenesis of SM and MCL. The two targeted therapies approved for MCL are midostaurin, a multikinase inhibitor, and avapritinib, a selective KIT D816V mutation-targeted tyrosine kinase inhibitor. Multiple drugs are being evaluated in clinical trials for managing MCL. MCL has a poor prognosis with a median overall survival (OS) of around 1.5 years. Further advancements and research are essential to develop treatments that may enhance median OS. In this article, we conducted a comprehensive yet simplified review of MCL, focusing primarily on its clinical manifestations and recent updates on management. We also identified the areas that require further research and emphasized the aggressive nature and poor prognosis associated with this disease.
Keywords: Mast cell leukemia; Systemic mastocytosis; Advanced systemic mastocytosis; KIT D816V mutation; Mast cell activation syndrome; Immunomodulatory therapy; Tyrosine kinase inhibitors; Midostaurin; Avapritinib; Hematopoietic stem cell transplantation; Overall survival
| Introduction | ▴Top |
Systemic mastocytosis (SM) is a rare disorder characterized by the abnormal proliferation, accumulation, and dysregulation of atypical mast cells (MCs) in various body organs, particularly in the bone marrow, liver, spleen, lymph nodes, and gastrointestinal (GI) tract. There are several subtypes of SM, which are classified based on the clinical features, histopathology, and molecular abnormalities [1-4]. Mast cell leukemia (MCL) is an exceedingly rare and aggressive subtype of SM characterized by the presence of > 20% atypical or immature MCs in the bone marrow aspirate (Fig. 1). MCL accounts for about 1% of SM cases and has an estimated prevalence of approximately 0.01 - 0.1 cases per 100,000 people. In this article, we review the subtypes, clinical features, molecular genetics, diagnosis, treatment, and prognosis of MCL in detail, with the goal of increasing the awareness of MCL among clinicians and contributing to the very limited literature available on this extremely rare disease. Understanding SM is essential for comprehending the pathophysiology and complications of MCL, which is also briefly reviewed here.
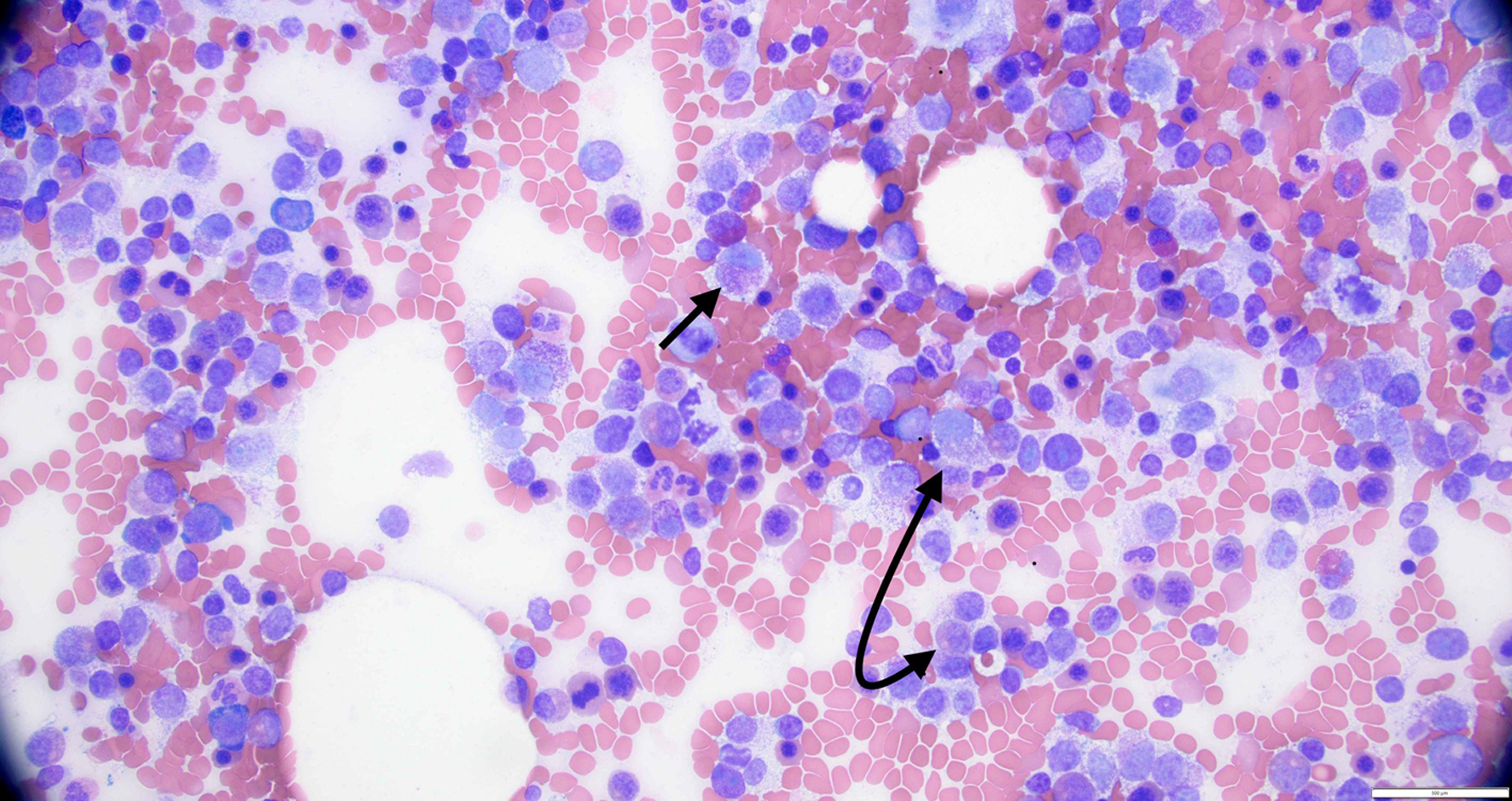 Click for large image |
Figure 1. High power view of bone marrow biopsy showing > 20% atypical mast cells (black arrows). |
| Classification of Mastocytosis | ▴Top |
The World Health Organization (WHO) classification and International Consensus Classification (ICC) are commonly used for the classification of mastocytosis [1, 2, 5]. Mastocytosis is broadly classified into three categories: cutaneous mastocytosis (CM), SM, and MC sarcoma (Table 1) [1-6].
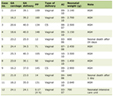 Click to view |
Table 1. Classification of Mastocytosis
(WHO/ICC) |
SM
According to the WHO classification, SM is diagnosed using either one major criterion plus one minor criterion or three minor criteria [1-5].
Major criterion is multifocal dense aggregates of > 15% MCs in the bone marrow biopsy or in the biopsy of an extracutaneous organ.
Minor criteria include: 1) > 25% of MCs in bone marrow biopsy or extracutaneous organ biopsy are either spindle-shaped or atypical/immature; 2) detection of any kind of receptor tyrosine kinase type III (KIT) mutation in the bone marrow, blood or extracutaneous organ; 3) detection of MCs expressing cluster differentiation (CD)25 with or without CD2 or CD30 in the bone marrow, blood, or extracutaneous organs; and 4) persistently elevated serum tryptase levels > 20 µg/L.
B-findings and C-findings are used to classify SM into further subtypes. B-findings are indicative of high MC burden in the body without the presence of organ damage. C-findings indicate the presence of organ damage from high MC burden.
B-findings include: 1) more than 30% of MCs in the bone marrow; 2) serum tryptase levels higher than 200 µg/L (normal range ≤ 10.9 µg/L); 3) palpable hepatomegaly or splenomegaly, or lymphadenopathy that can be palpated or reported in an imaging study; and 4) findings of myelodysplasia in non-MC lineages, but not meeting criteria to diagnose as an associated hematopoietic neoplasm.
Examples of C-findings include: 1) cytopenias (leukopenia, anemia, and thrombocytopenia) on bone marrow biopsy; 2) osteolytic lesions or pathologic fractures of the bones; 3) malabsorption and GI bleeding; and 4) hepatomegaly with transaminitis and splenomegaly with hypersplenism.
SM subtypes
SM is divided into six subtypes based on the disease characteristics (Table 2): 1) indolent systemic mastocytosis (ISM); 2) bone marrow mastocytosis (BMM); 3) smoldering systemic mastocytosis (SSM); 4) systemic mastocytosis with an associated hematologic neoplasm (SM-AHN); 5) aggressive systemic mastocytosis (ASM); and 6) MCL.
Click to view |
Table 2. Brief Overview of Systemic
Mastocytosis Subtypes |
The three variants SM-AHN, ASM, and MCL are classified as advanced systemic mastocytosis (AdvSM), which indicates a poor prognosis [7, 8].
| Epidemiology of MCL | ▴Top |
SM itself has an estimated prevalence of around 1 - 10 cases per 100,000 individuals. MCL is a rare and aggressive subtype, constituting around 1% of all SM cases. MCL accounts for less than 1% of all leukemias, making it one of the rarest hematologic malignancies [1, 8-10]. MCL typically affects adults, with a median age at diagnosis ranging from 40 to 60 years, although cases have been reported in both younger and older populations. There appears to be a slight female dominance with a male-to-female ratio of 1:1.5 [1, 9, 10]. Pertinent comorbidities associated with mastocytosis include immunoglobulin E (IgE)-dependent allergies, vitamin D deficiency, and psychiatric or psychological conditions, which have no direct causative effect [2].
MCL itself is further classified as: 1) Primary MCL (occurring de novo without prior mast cell disorders) or secondary MCL (arising from a preexisting MC neoplasm or SM). Primary MCL is more common (60-70% of cases) and aggressive with a poor prognosis than secondary MCL [8-10]. 2) Acute MCL (60-90% of patients) that is aggressive with C-findings that indicate organ damage, mainly bone marrow dysfunction (typically cytopenia), hepatosplenomegaly with liver failure (transaminitis, ascites, and portal hypertension) and hypersplenism, skeletal involvement (osteoporosis, big osteolytic lesions, osteoblastic lesions, and pathological fractures) and GI tract (diarrhea, malabsorption, and failure to thrive) or chronic MCL that is indolent without C-findings [8-10]. Histopathologically, this is driven by the immaturity of MCs. Chronic MCL has more mature MCs, ranging from atypical spindle-shaped MCs to MCs resembling normal MCs, and acute MCL typically presents with abundant immature MCs, which resemble metachromatically granulated blast cells [11]. 3) Less common leukemic MCL (> 10% circulating atypical MCs in peripheral blood) and more common aleukemic MCL (< 10% circulating atypical MCs in peripheral blood; constitutes 60-65% of all cases of MCL) [8-10]. 4) More common MCL alone and less common MCL with AHN (about 30% of patients). Common AHN associated with MCL is myelodysplastic neoplasm (MDS)/myeloproliferative neoplasm (MPN)-unclassifiable (about 45%), followed by chronic myelomonocytic leukemia (CMML) (about 25%), chronic eosinophilic leukemia (about 10%), and acute myeloid leukemia (AML) (about 10%) [9, 10]. Non-Hodgkin lymphoma (NHL) and plasma cell myeloma are seen in about 3% of cases with MCL-AHN, and these patients do not have KIT mutations.
| Pathophysiology, Immunophenotyping, and Molecular Genetics of MCL | ▴Top |
MCs are essential in inflammatory and immune responses like innate and acquired immunity, allergic mechanisms, wound healing, coagulation pathways, and autoimmune conditions. MCs have a greater lifespan (months) compared to the other leucocytes and usually do not circulate in the peripheral blood. Bone marrow hematopoietic cells that differentiate into MCs generally express CD2, CD13, CD25, CD30, CD33, CD44, and CD117. Common mediators released by MCs are histamine, tryptase, heparin, leukotrienes, prostaglandins, and cytokines. These mediators play an important role in MC activation syndrome. CD117 and tryptase are mainly used to diagnose MCL by detecting MC infiltrates in the bone marrow (Figs. 2 and 3) and other tissue (mainly bone) biopsies (Figs. 4-6). Another sensitive marker for MCL is CD25 [12, 13].
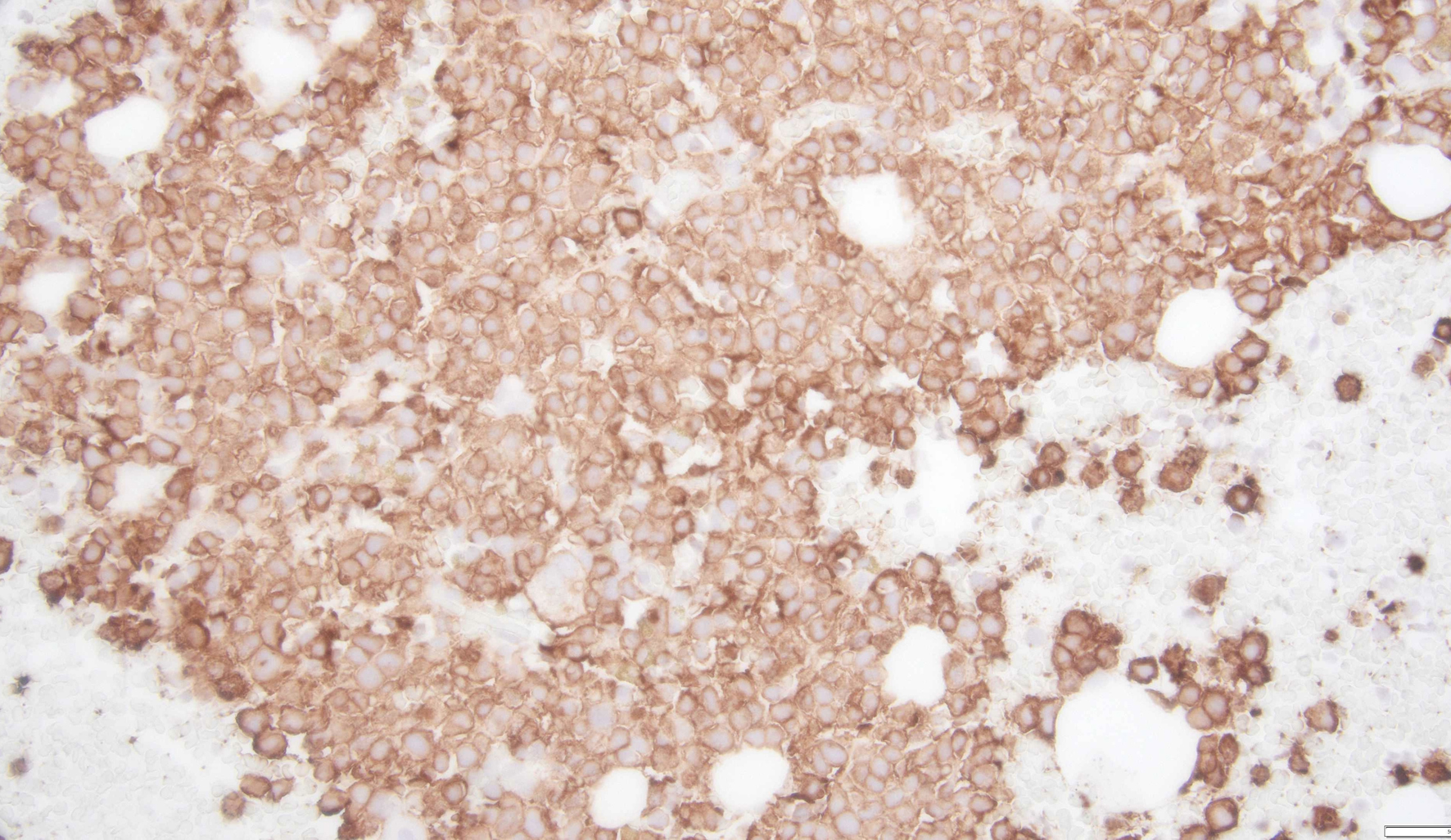 Click for large image |
Figure 2. A clot section of bone marrow biopsy showing dense infiltration and clustering of atypical mast cells with diffuse and increased CD117 expression. |
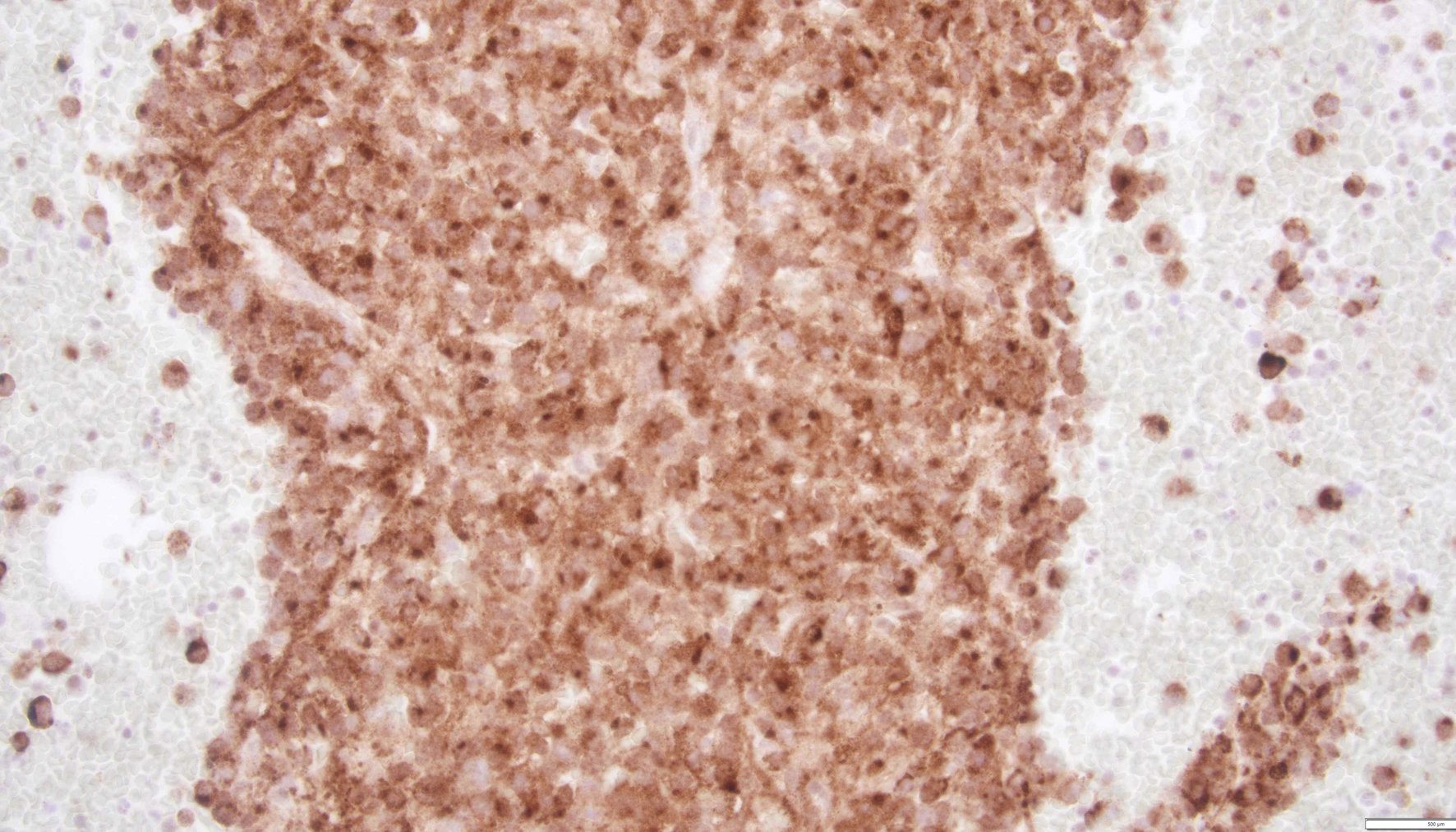 Click for large image |
Figure 3. A clot section of bone marrow biopsy with dense infiltration and clustering of atypical mast cells with diffuse tryptase staining. |
 Click for large image |
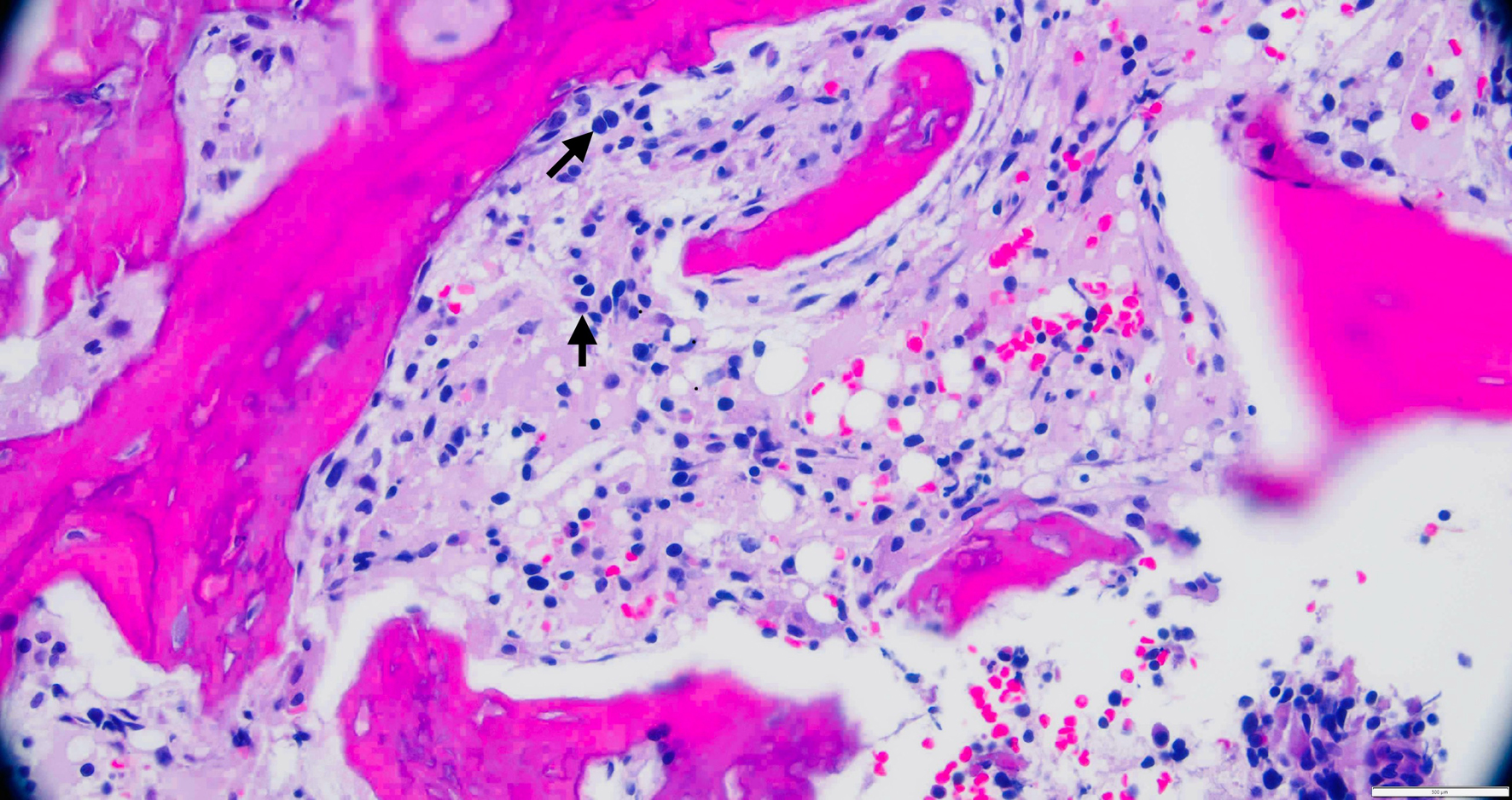
Figure 4. A high-power view of an iliac bone biopsy showing an increased number of atypical mast cells (black arrows). |
 Click for large image |
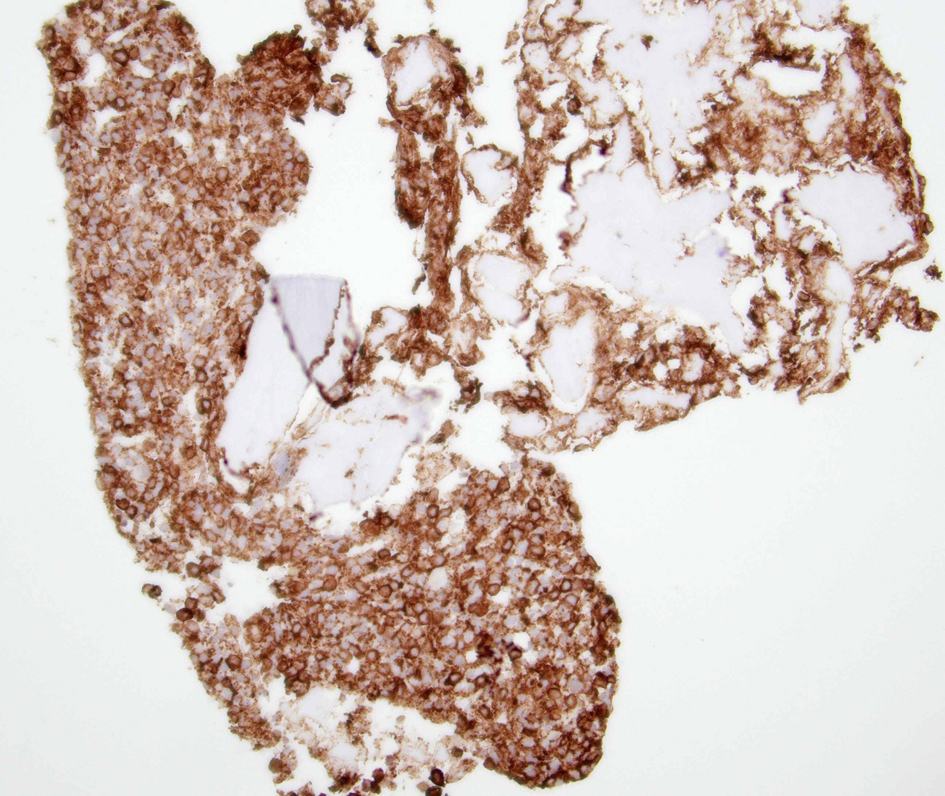
Figure 5. A clot section of the iliac bone biopsy showing cells with diffuse CD117 expression. |
 Click for large image |
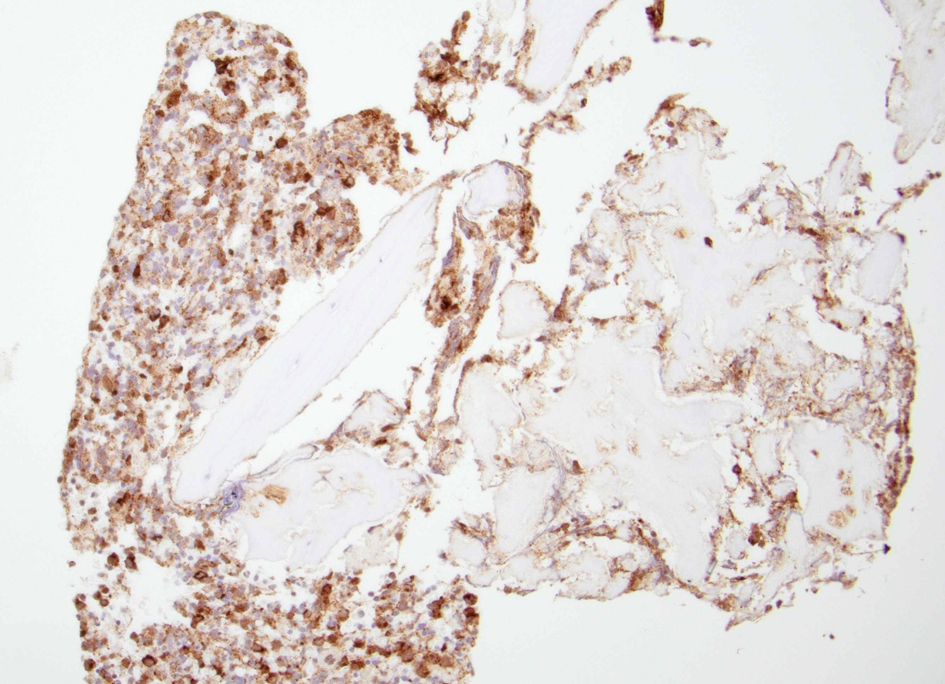
Figure 6. A clot section of the iliac bone biopsy showing an increased number of cells with diffuse tryptase staining. |
The most common molecular abnormality observed in MCL is the mutations in the KIT gene [14-16]. KIT encodes a receptor tyrosine kinase that plays an important role in MC growth, differentiation, and proliferation. After interacting with the stem cell factor, KIT activates the downstream pathways, including multiple tyrosine kinases, mainly P13K, Janus tyrosine kinase/signal transducers and activators of transcription (JAK/STAT), and rat sarcoma (RAS)/extracellular signal-regulated kinase (ERK) [15, 16]. A mutation in KIT often results in constitutive activation of the KIT receptor, leading to uncontrolled proliferation and extended survival of mast cells. Among the mutations in KIT, the D816V mutation is particularly prevalent in MCL and other types of SM, present in approximately 65% of cases of MCL [15, 16]. Another 20-25% of MCL cases harbor D816H or D816Y mutations; hence, 90% of cases with MCL harbor some kind of KIT D816 mutation [9, 17, 18].
Other genetic abnormalities have been reported in MCL, although they are less common and poorly characterized. These include mutations in genes such as TP53 R273H, ten-eleven translocation (TET)2, additional sex combs-like (ASXL)1, serine and arginine rich splicing factor 2 (SRSF2), Casitas B-lineage lymphoma (CBL), RAS, and runt-related transcription factor 1(RUNX1), which are commonly associated with myeloid neoplasms [12, 17, 19]. Mutations in one of these genes are present in approximately 50% of MCL cases and usually indicate a worse prognosis [18]. Rarely, in some cases of SM/MCL-AHN, the JAK2 V617F mutation was found in conjunction with the KIT D816V mutation [9, 20].
Cytogenetic studies have identified various chromosomal abnormalities in MCL; however, there is no specific recurring abnormality that is characteristic of this disease. Some of the reported abnormalities include trisomy 8, deletion of chromosome 5q, and deletion of chromosome 7q [17, 18]. Epigenetic dysregulation, including alterations in DNA methylation and histone modifications, may also contribute to the pathogenesis of MCL [17, 18]. These epigenetic changes can affect gene expression patterns, including those involved in cell proliferation, survival, and differentiation. Limited data are available on chromosomal abnormalities and epigenetic dysregulations in MCL.
Understanding the molecular genetics of MCL is crucial for developing targeted therapies and personalized treatment approaches. Since the implication of KIT mutation in the pathogenesis of SM and the development of tyrosine kinase inhibitors (TKIs), the management of SM has significantly advanced. Previous reports have shown that only rare KIT D816V negative cases were responsive to the initial generation of TKIs like imatinib and dasatinib [21]. But the development of newer TKIs like midostaurin and avapritinib, which have activity against KIT D816V, has increased the treatment prospects of SM and hence MCL [12, 22].
| Clinical Manifestations of MCL | ▴Top |
Clinical manifestations of MCL are defined by the type and extent of organ involvement [8-10]. Clinical features in chronic MCL without C-findings are mostly limited to cutaneous findings, organomegaly (without dysfunction), lymphadenopathy, and constitutional symptoms. Clinical characteristics are broadly divided into three categories: organ dysfunction [3, 8-11] (C-findings), constitutional, and mast cell activation syndrome (MCAS) (Table 3).
Click to view |
Table 3. Clinical Features and Organ
Dysfunctions in MCL |
Organ dysfunction (seen in acute MCL)
Skin: blistering lesions
Skin lesions are present in secondary MCL and generally absent in primary MCL.
GI tract
Abdominal pain, diarrhea, bleeding from peptic ulcer disease, and malabsorption.
Musculoskeletal
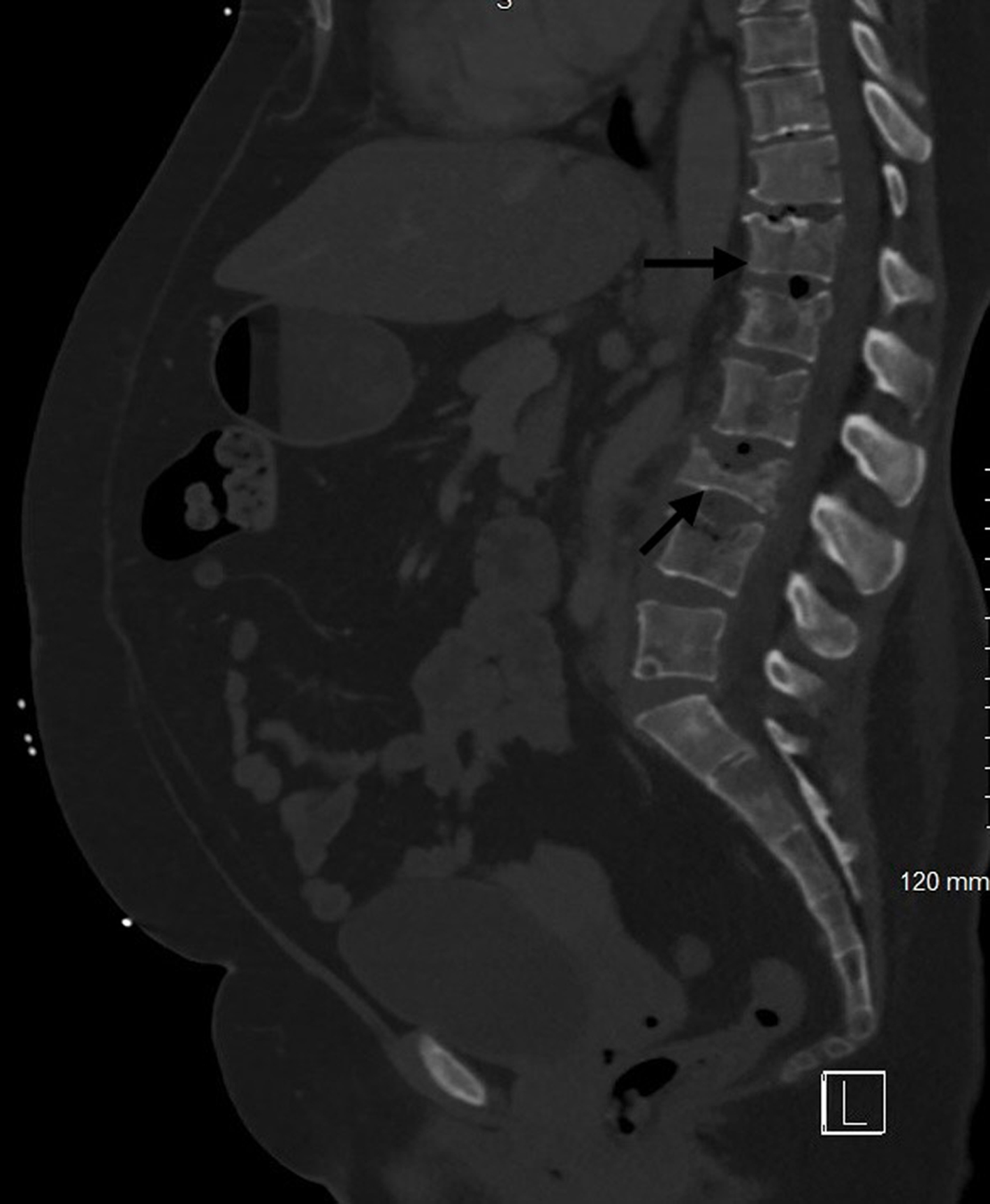
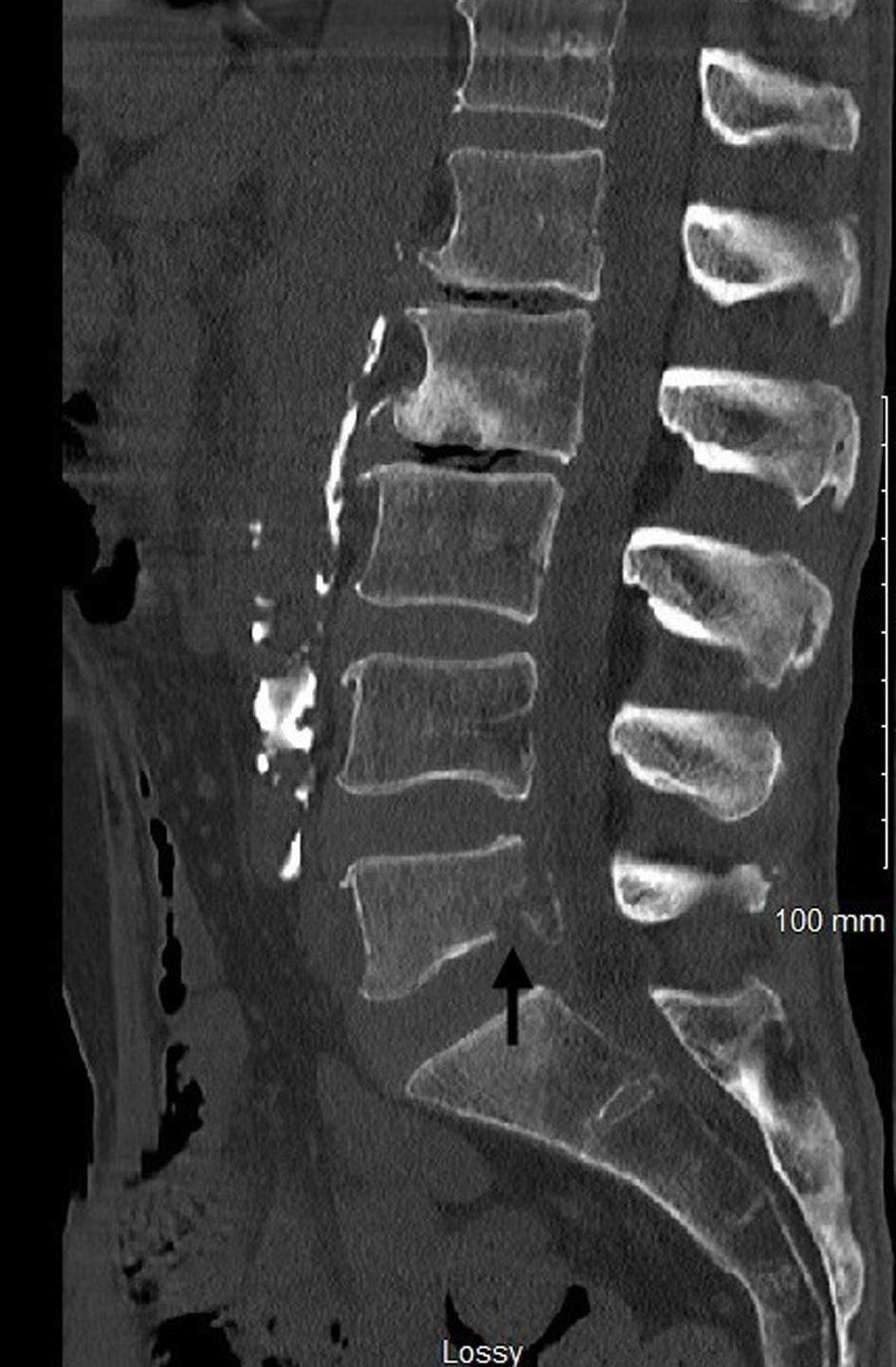
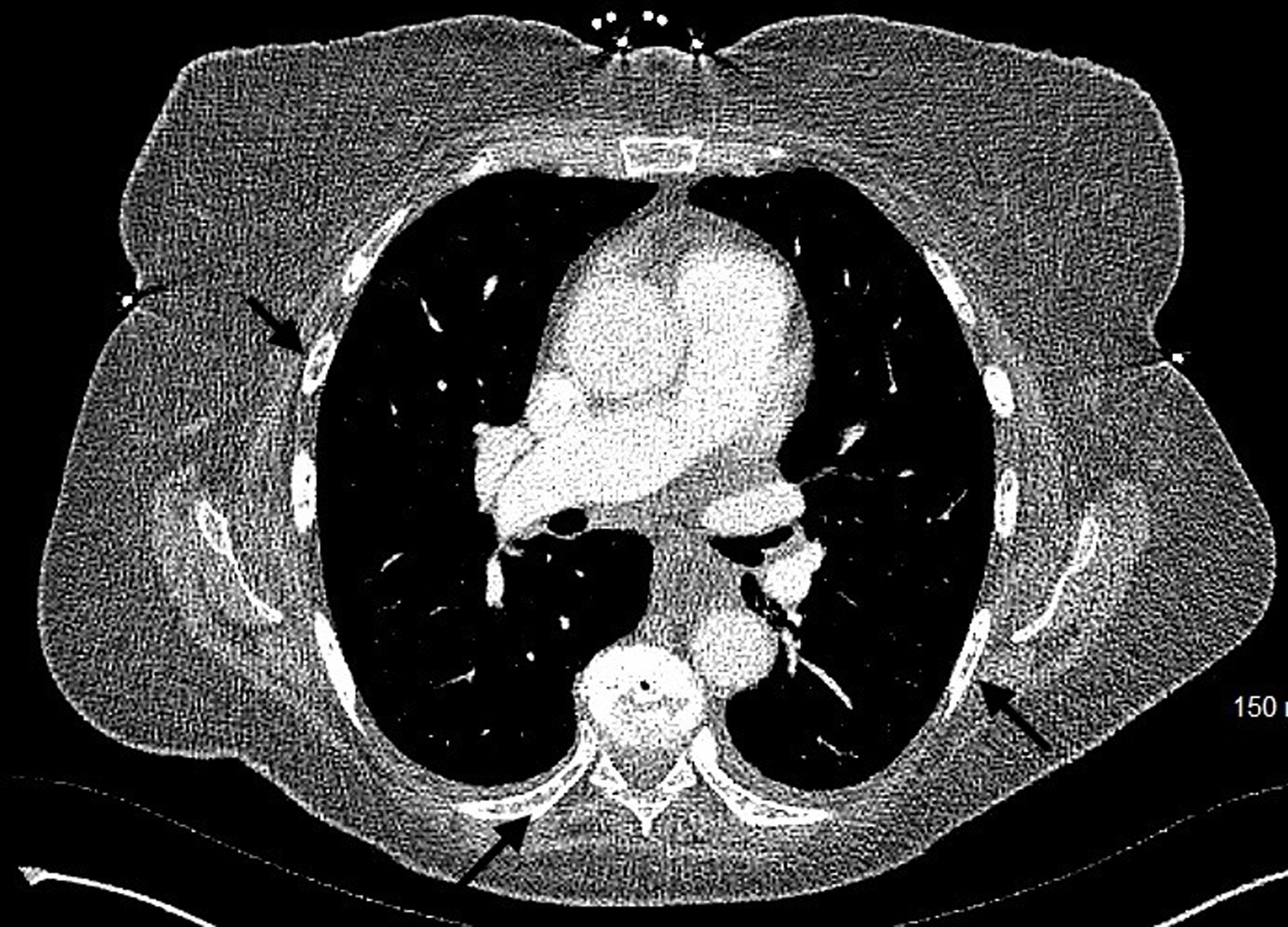
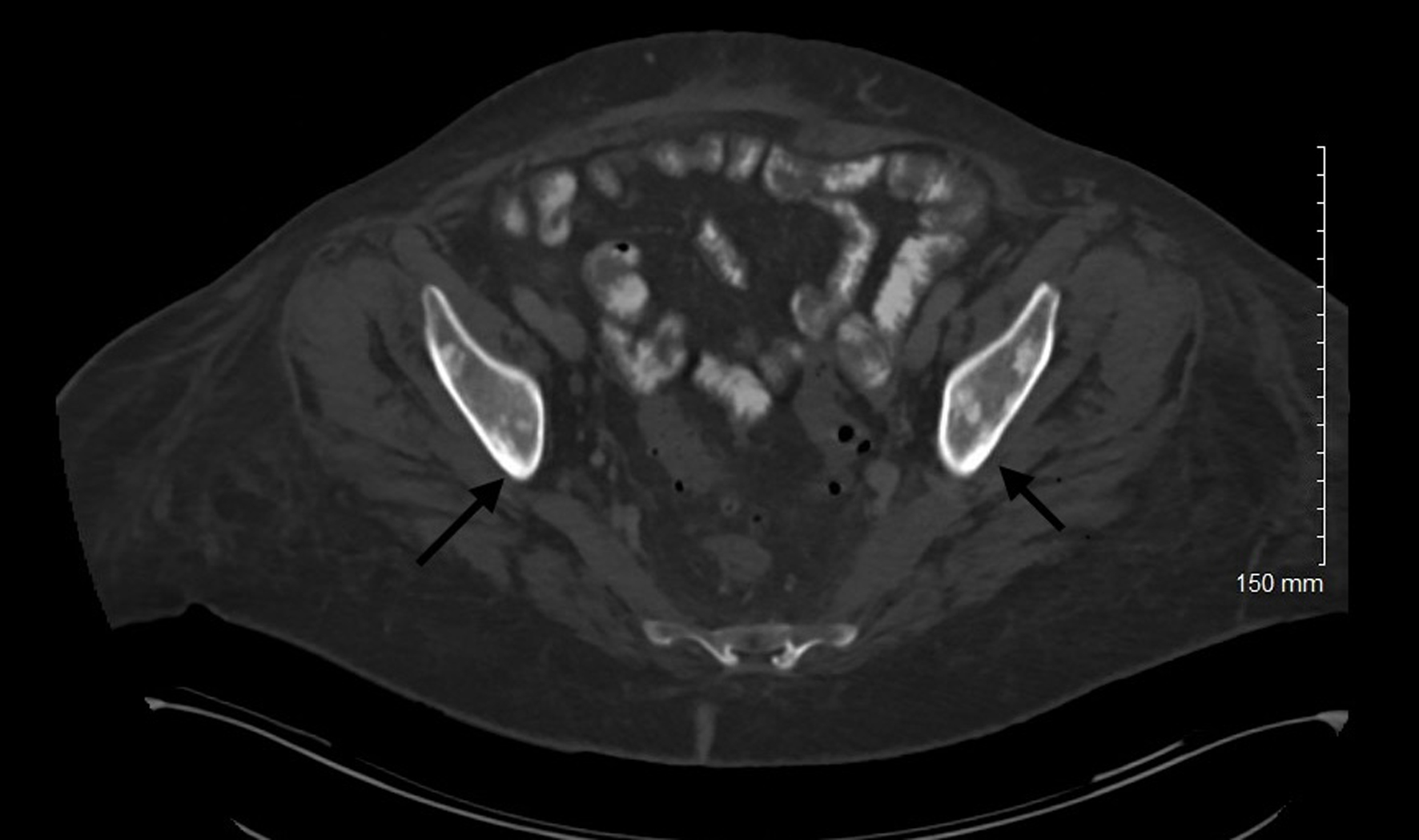
Bone involvement is common in MCL and has both osteolytic and osteoblastic lesions. Patients present with musculoskeletal pain and physical deconditioning. Typical presentation is osteoporosis and focal osteolytic lesions with pathological fractures (Figs. 7 and 8). Osteoblastic (sclerotic) lesions are not common (Figs. 9 and 10) [23-25]. It is observed that the bone metastases outside of the vertebral column are uncommon, and lesions in the appendicular skeleton are extremely rare [23-25] (Fig. 11).
 Click for large image |
Figure 7. Computed tomography of lumbar spine with contrast showing the osteolytic lesions and pathological fractures in multiple lumbar vertebrae (black arrows). |
 Click for large image |
Figure 8. Computed tomography of lumbar spine with contrast showing the osteolytic lesion with fracture in the L5 lumbar vertebra (black arrow). |
 Click for large image |
Figure 9. Computed tomography of chest with contrast showing the osteoblastic lesions in multiple ribs (black arrows). |
 Click for large image |
Figure 10. Computed tomography of spine with contrast showing the osteoblastic lesions in the sternum and multiple thoracic and lumbar vertebrae (black arrows). |
 Click for large image |
Figure 11. Computed tomography of abdomen and pelvis with contrast showing osteoblastic lesions in the pelvis (appendicular skeleton) (black arrows). |
Bone marrow
Pancytopenia (anemia, thrombocytopenia, and leukopenia), immunosuppression with infections leading to sepsis.
Central nervous system (CNS)
Headache, dizziness, cognitive impairment, and neuropsychiatric symptoms.
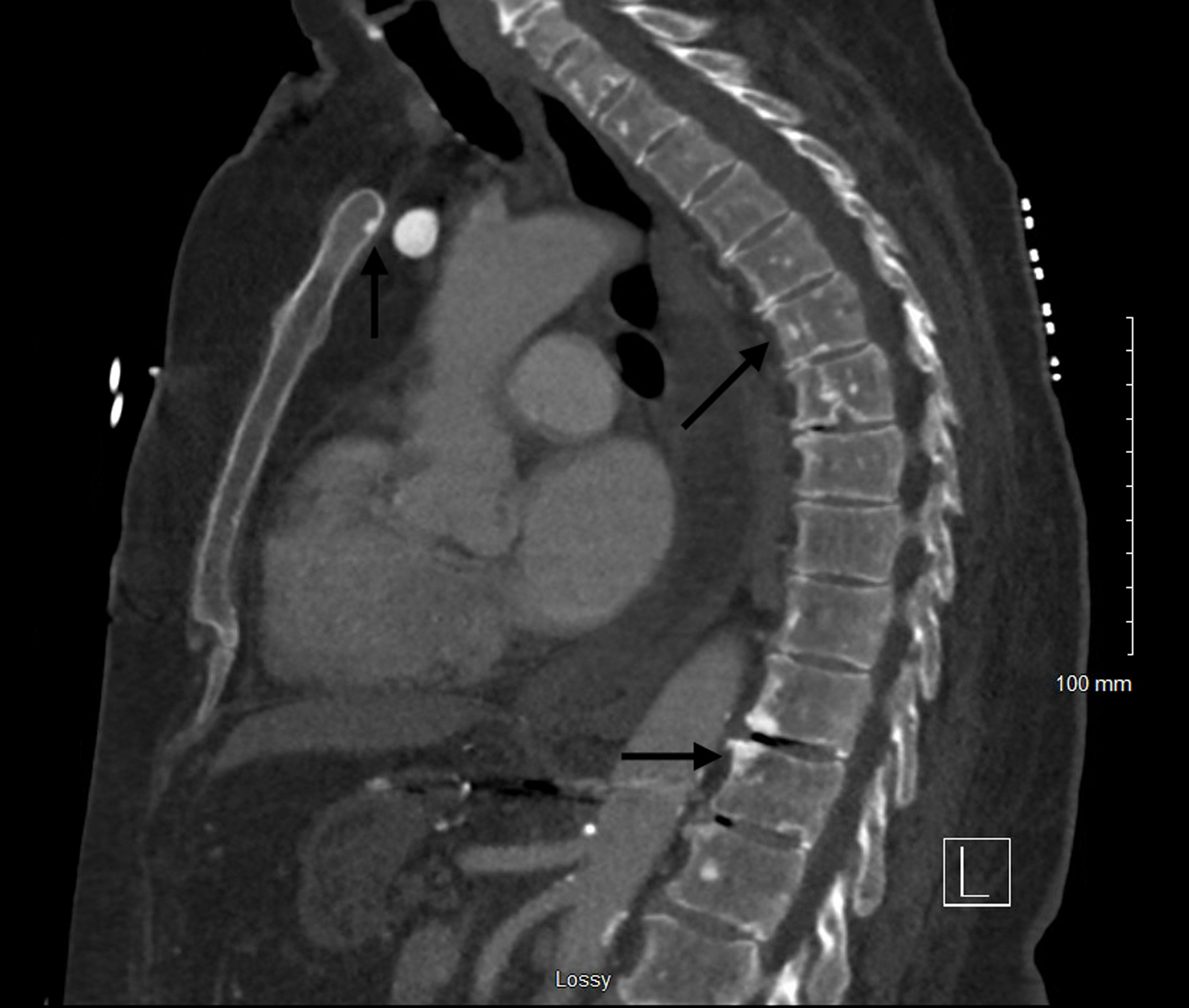
Hepatomegaly with hepatic dysfunction
Transaminitis, hypoalbuminemia, coagulopathy including disseminated intravascular coagulation (DIC), ascites, and portal hypertension (Fig. 12) [25].
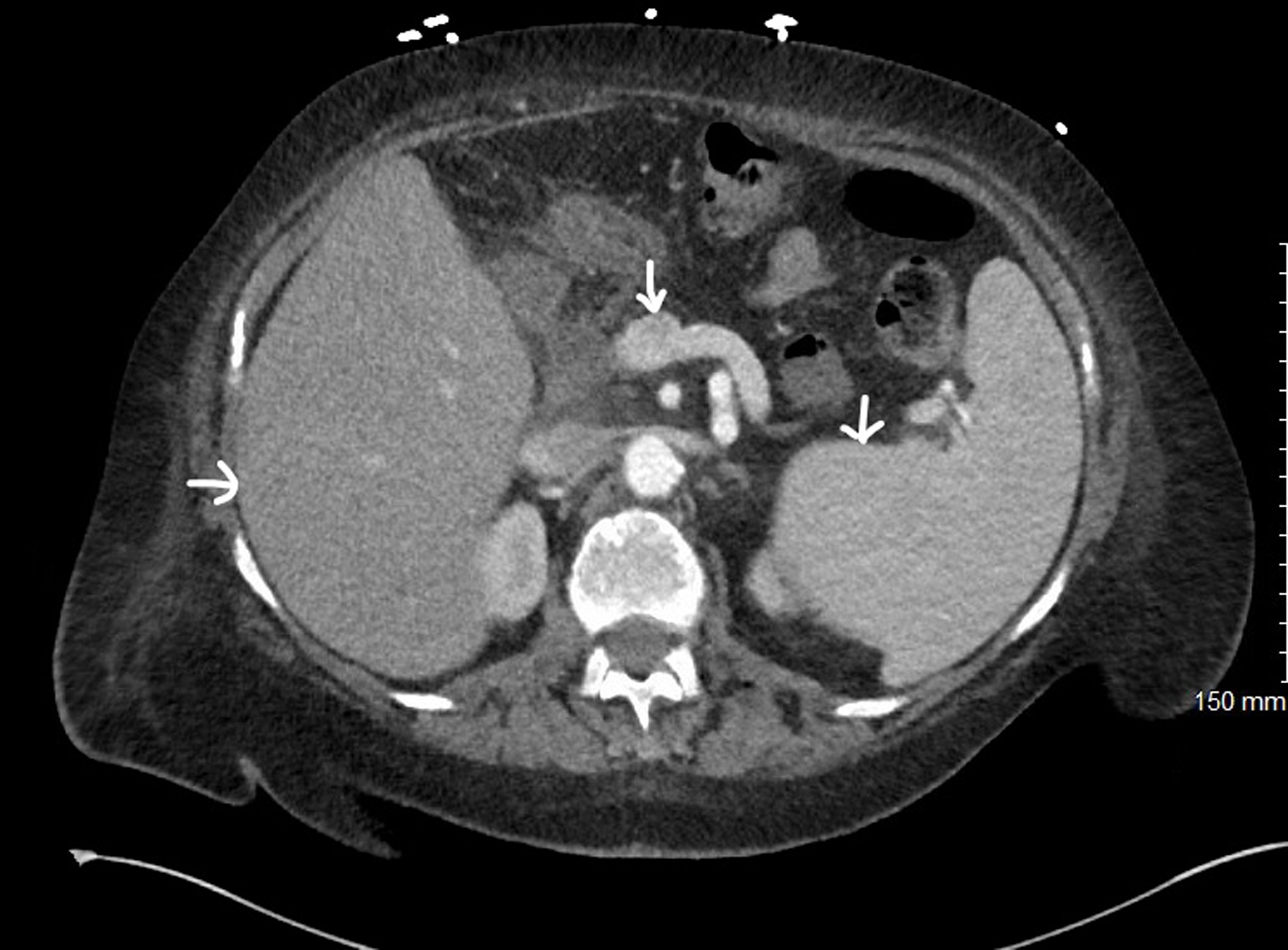 Click for large image |
Figure 12. Computed tomography of abdomen and pelvis with contrast showing hepatosplenomegaly and portal vein dilatation (white arrows). |
Splenomegaly with hypersplenism
Left-sided abdominal pain and fullness, early satiety, and one or more cytopenias (Fig. 12).
Diffuse lymphadenopathy
Enlargement of lymph nodes due to MC infiltration. It could be painful.
Constitutional symptoms: common presentations in chronic MCL
Constitutional symptoms include fever, fatigue, anorexia, malaise, decreased exercise tolerance, weight loss, and night sweats.
MCAS
In some cases, this could be the presenting illness and can manifest in any type or stage of MCL. These findings correlate with those of anaphylaxis. The best marker in serum for MCAS is elevated tryptase levels [26]. It includes: 1) cardiovascular system: hypotension/shock, tachycardia, and syncope; 2) cutaneous: pruritus, hives, urticaria, angioedema, and flushing; 3) respiratory system: wheezing, shortness of breath, and stridor; and 4) GI tract: diarrhea, nausea, vomiting, bloating, early satiety, and abdominal pain.
In many cases, patients present with nonspecific symptoms like fever, fatigue, skin allergies, or musculoskeletal pain, and only after further probing might we see one or more of the above-mentioned findings. It is important to note that MCL can be rapidly progressive and life-threatening, often leading to multi-organ failure if left untreated. In many patients, the complications that arise from the SM component require urgent intervention than the MCL itself [8, 10].
| Diagnostic Evaluation of MCL | ▴Top |
The workup of MCL typically involves bone marrow biopsy, peripheral blood smear examination, and assessment of serum tryptase levels. Immunohistochemistry and flow cytometry are used to detect abnormal MC populations. The diagnosis of MCL relies on bone marrow biopsy demonstrating significant infiltration of MCs with atypical morphology (Fig. 1) and expression of specific markers such as CD2, CD25, and CD30 in addition to typical MC markers like CD117 and tryptase (Figs. 2 and 3) [9, 16, 18]. In some cases, bone biopsy of the osteolytic or osteoblastic lesions showing similar MC findings and markers aid in the diagnosis (Figs. 4-6). Immunophenotyping and genetic markers are described in detail above.
Common laboratory studies that will help in the management of MCL include complete blood count (CBC), basic metabolic panel (BMP), coagulation panel, and liver function tests (LFTs). Abnormalities in these tests suggest various organ dysfunctions, notably bone marrow/spleen (cytopenias), liver (transaminitis, coagulopathy, and hypoalbuminemia), and GI tract (anemia, electrolyte deficiencies). Imaging studies like computed tomography (CT) scans and magnetic resonance imaging (MRI) studies will show hepatosplenomegaly, portal hypertension (Fig. 12), lymphadenopathy, and bone metastases (Figs. 7-11). Reverse transcription-polymerase chain reaction (RT-PCR) plus restriction fragment length polymorphism (RFLP), PCR plus Sanger sequencing, digital PCR (dPCR), and next-generation sequencing are some of the commonly used methods to detect KIT D816 mutations [9, 10, 16-19].
| Management of MCL | ▴Top |
Currently, treatment options for MCL remain limited and the prognosis is generally poor, highlighting the need for further research into treatments targeting the underlying molecular mechanisms. Chemotherapy regimens used for other types of leukemias are mostly ineffective against AdvSM/MCL, necessitating the exploration of alternative strategies. Targeted therapies, such as TKIs and monoclonal antibodies aimed at KIT mutations, have shown promising results; however, the heterogeneity of AdvSM and the lack of multiple specific molecular targets pose challenges in developing effective treatments. Numerous studies and ongoing clinical trials are evaluating targeted therapies for managing AdvSM/MCL. We have categorically summarized various treatment modalities available and reviewed some of the pertinent studies and clinical trials in the process (Table 4, Fig. 13).
Click to view |
Table 4. Summary of Common Drugs Used to Manage
MCL and Their Notable Adverse Effects |
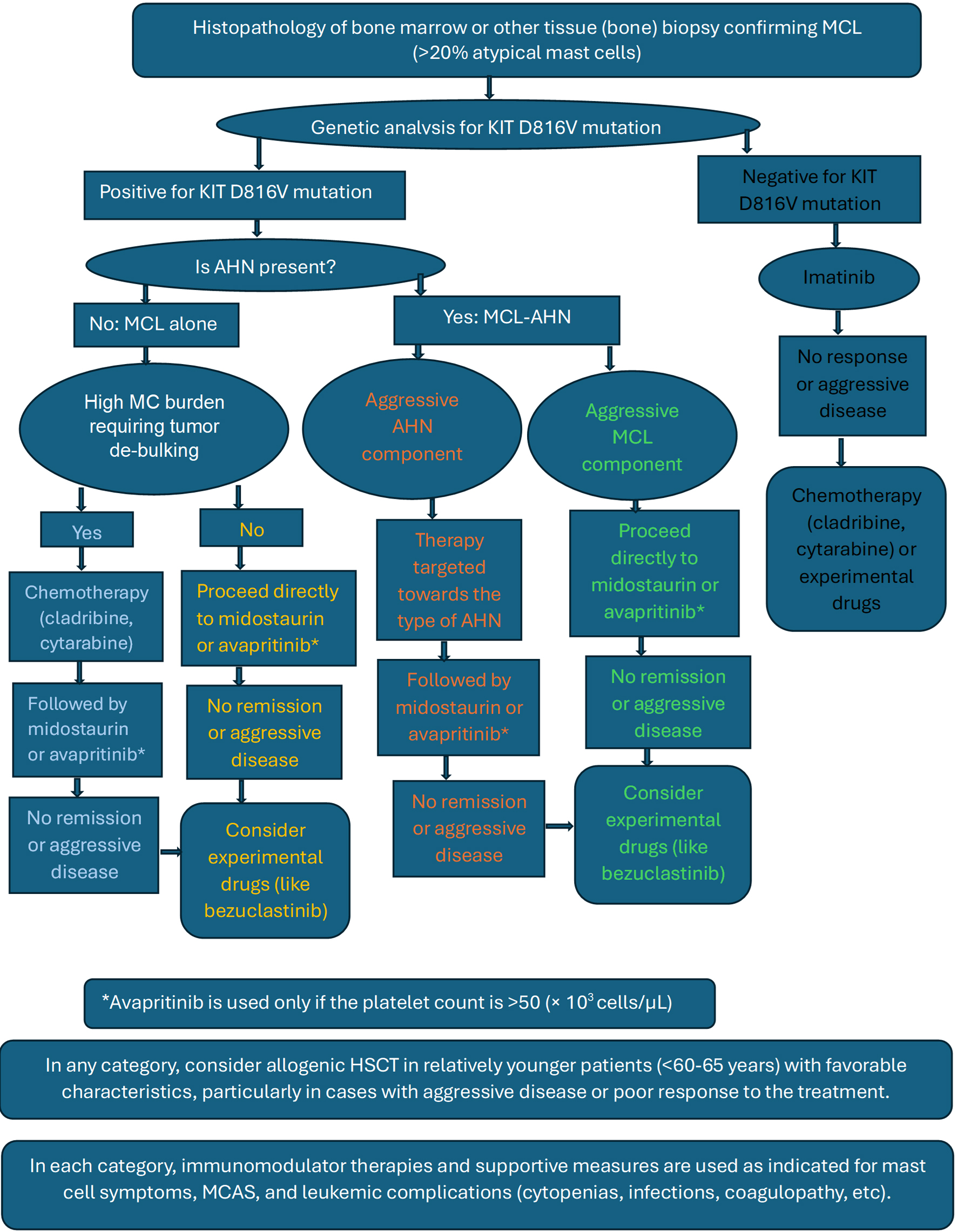 Click for large image |
Figure 13. Basic treatment algorithm recommended for mast cell leukemia. |
Targeted therapy
In AdvSM, including MCL, targeted therapies using TKIs are considered as first-line therapy. The two targeted therapies approved for MCL are midostaurin and avapritinib [8-10, 22, 27-29].
Midostaurin, a multikinase inhibitor, has demonstrated efficacy in KIT D816V-positive AdvSM. Several studies have reported that midostaurin improves prognosis regardless of the disease stage [22, 27-29]. The phase II trial by Gotlib et al was crucial in approving midostaurin for patients with advanced SM, including highly fatal variants of MCL [30]. Data analysis from these studies showed an estimated overall response rate (ORR) of 20-38% and a median OS of 12.2 months, indicating a doubled survival rate with midostaurin use compared to the historical report of 6 months. Complete remission was rare, short-lived, and occurred in less than 10% of patients.
In a phase IV clinical trial and literature review, researchers reported that imatinib mesylate (a multitype TKI that targets BCR-ABL) was effective in SM patients without the KIT D816V mutation, with some patients even achieving a complete response. However, imatinib was ineffective in patients with the KIT D816V mutation [21].
Dasatinib, an SRC/ABL inhibitor with activity against imatinib-resistant BCR-ABL forms and KIT D816V mutation, showed promising results in various types of SM in vitro but showed limited results when tried clinically. In a phase II study, authors reported that dasatinib therapy helped control the disease symptoms, but neither did it eliminate the disease, nor were its side effects desirable when used in a selected group of SM patients with KIT D816V mutation [31].
Nilotinib (a second-generation BCR-ABL TKI) had only a minimal response, even when used in selective cases of AdvSM, and masitinib (c-KIT TKI) was hardly ever tested in AdvSM [8, 28].
Two clinical trials, which assessed ibrutinib (a Bruton’s tyrosine kinase (BTK) inhibitor targeting the B-cell receptor pathway) in patients with AdvSM, were discontinued due to no proven benefit [32, 33].
Avapritinib, a selective KIT D816V mutation-targeted TKI, is approved in patients with advanced SM and MCL. Results from two studies, the Explorer phase I trial [34] and the Pathfinder phase II trial [35], showed that avapritinib has a good response and a favorable benefit-to-risk profile in patients with AdvSM, irrespective of prior treatment or disease subtype. These studies led to the Food and Drug Administration (FDA) approval of avapritinib in AdvSM, including MCL. These studies reported an ORR and a 2-year OS of 73-75% and 75-79%, respectively, for all patients with AdvSM. Lower rates were observed in the MCL subgroup, with an approximate ORR of 50% and a median OS of 9 months. The Pioneer study evaluated the safety and efficacy of avapritinib in ISM, and results from the study led to the FDA approval of avapritinib in ISM. The study showed that avapritinib reduced tryptase levels by > 50% in more than half of the patients in 6 months [36].
Chemotherapy
Chemotherapy regimens commonly used in treating AML are often employed in MCL, due to similarities in disease biology and clinical behavior. These chemotherapeutic agents include cladribine (a purine analogue, 2-CdA), interferon-alpha (IFN-α), anthracycline-based regimens (such as cytarabine and daunorubicin), and fludarabine [8, 29, 37-39].
Cladribine
The effectiveness of 2-CdA as an initial therapy for controlling MC burden before establishing definitive treatments is strongly supported by multiple studies [37-39].
Helbig et al concluded in their study that 2-CdA appears beneficial in some cases of AdvSM, but the response was noted to be incomplete and transient. No complete responses were demonstrated, particularly in patients with MCL [40].
Current evidence suggests 2-CdA use in patients unfit for TKI or in KIT D816V-negative disease. It has an estimated ORR of 10-30% and a median OS of 4-6 months [12, 37-39].
Cytarabine
Cytarabine use in MCL has been reported only in limited case reports and small retrospective series. It is not a standard treatment and has been mainly used in combination with other treatments in patients with aggressive MCL or as a bridge to transplant or experimental therapies. It has an estimated ORR of < 20% and a median OS of 4 - 6 months [37, 38, 40].
For younger patients where hematopoietic stem cell transplantation (HSCT) is considered, debulking is done before HSCT with polychemotherapy, i.e., high doses of cytarabine and fludarabine [40].
Valent et al in their studies demonstrated that the patients with SM, including AdvSM/MCL, showed a response to chemotherapy with cytarabine and 2-CdA but concluded that these results need to be confirmed by larger trials [37, 38].
IFN
IFN-alpha-2b plus prednisolone in a smaller study had beneficial effects when used in a subgroup of patients with AdvSM, particularly in the early phases of the disease. However, the response was transient in MCL, which required more aggressive therapy [41].
A clinical trial designed to assess the efficacy of combination therapy using 2-CdA and pegylated IFN-alpha-2a in patients with AdvSM demonstrated no notable efficacy and hence was discontinued [42].
Immunomodulatory therapy
The main role of immunomodulatory therapy in MCL is to control MC-mediated symptoms and support the immune response, mainly in chronic MCL. It is important to note that it is not curative and is generally used in conjunction with targeted treatments or chemotherapy. These agents include thalidomide, lenalidomide, cromolyn, corticosteroids, and interleukin-2 (IL-2) [8, 9, 43, 44].
Thalidomide (an anti-inflammatory and anti-angiogenic drug) is used in SM to control MC-related symptoms. In a multicentric phase II study, authors reported that thalidomide is an acceptable and effective treatment in AdvSM when used in selected patients, considering the risk vs. benefit profile [43]. The study also showed that thalidomide achieved longer remissions compared to IFN-alpha when used in the benign forms of SM, but its use in AdvSM/MCL is not well studied [43].
A multicenter randomized trial reported potential improvement in MC-related symptoms and overall quality of life with the use of inhalation cromolyn (PA101) in ISM, but concluded that further clinical assessment in larger trials would be necessary to confirm the long-term benefits and safety of this approach in ISM and other SM subtypes [44]. Cromolyn is commonly used in severe cases of MAS (irrespective of the SM or MCL subtype).
Corticosteroids are frequently used to reduce the inflammatory response associated with SM/MCL and can alleviate symptoms like skin rash, GI issues, bone pain, and anaphylaxis by reducing MC mediator release. However, the role of corticosteroids is only short-term symptom control, and they do not modify the disease course [10, 12, 41]. Long-term use of steroids may lead to significant, commonly known side effects.
HSCT
Allogenic HSCT (allo-HSCT) is considered for eligible patients, particularly those with high-risk disease, multi-mutated pattern, or limited remission with induction chemotherapy or targeted therapies [29, 40, 45]. HSCT is also a viable treatment alternative for patients with relapse after chemotherapy and/or targeted therapies and a negative KIT D816V mutation [46]. HSCT remains the mainstay of treatment in younger patients with compatible donors. Chemotherapeutic agents or targeted therapies may be utilized before and after HSCT to optimize outcomes and improve OS [40, 46].
In a study, authors reported that allo-HSCT improves long-term survival and OS in patients with advSM, including MCL. The study also highlighted the importance of patient selection and pre-transplant conditioning of the selected patients. A complete response was noted in approximately 27% of patients, with a median OS of 11 months [40].
Autologous HSCT is generally less favored in MCL due to limited evidence of efficacy. Since autologous HSCT reinfuses the patient’s own cells, there’s a risk of reintroducing malignant MCs [40, 45].
Graft-versus-MCL effect: There is some evidence suggesting that the graft-versus-leukemia effect could contribute to disease control post-HSCT, as the new immune cells from the donor may help eliminate residual malignant MCs. The existence of this effect was studied in MCL by Spyridonidis et al in 2004 [47]. This study reported that allo-HSCT reduced more aggressive forms of SM to less benign, indolent forms and thus improved overall prognosis.
In a trial conducted by National Heart, Lung, and Blood Institute (NHLBI), it was concluded that HSCT might be a feasible treatment option for patients with AdvSM, particularly in those with severe systemic disease that is unresponsive to targeted therapy or who have associated hematologic neoplasms [48].
Allo-HSCT offers the potential for durable remissions but is associated with significant risks of transplant-related complications, including graft-versus-host disease (GvHD), infections, and increased mortality, which must be carefully managed, especially in the context of MCL [34, 40, 48]. Patient selection should be carefully weighed against the risks and benefits of transplantation, considering factors such as age, comorbidities, and disease status. Most studies recommended that allo-HSCT should be reserved for younger patients with good performance status due to the high risk of complications [40, 46].
Management of MCL-AHN
The critical and most crucial step is integrating and analyzing the clinical, histopathological, and molecular data to assess which disease component (MCL or AHN) needs urgent treatment [5, 8-10, 29].
In most patients, the MCL component requires urgent intervention, and treatment should be started with either midostaurin or avapritinib, with HSCT preserved for refractory cases. If the AHN component is aggressive, particularly in cases with stable forms of MCL and aggressive AHN like AML or poor risk CMML, treatment directed towards it should be started first (i.e., high-dose chemotherapy followed by HSCT), along with management of MC symptoms [5, 9, 10, 29].
Supportive care
Supportive measures play a crucial role in managing complications such as cytopenias, infections with sepsis, tumor lysis syndrome (TLS), organ dysfunction, and MCAS [8, 9, 34]. Blood product transfusion, hematopoietic growth factors, and antimicrobial treatment/prophylaxis should be considered as appropriate, but must be cautiously used to prevent additional treatment-related complications. Adequate hydration before and during treatment, pre-/post-treatment use of allopurinol or rasburicase, and gradual dosing of drugs are implemented to control the complications of TLS [9, 37].
Proton pump inhibitors, along with H2-blockers, are used in patients with gastrointestinal symptoms like gastroesophagitis or peptic ulcers. Corticosteroids are used for respiratory compromise with wheezing, musculoskeletal (bone) pain, and allergic symptoms [9, 46]. Osteoporosis with or without osteolytic lesions is managed with bisphosphonates, alendronate weekly or pamidronic acid/zoledronic acid weekly [5, 9, 46].
Treatment of MCAS in MCL can be achieved by blockade of mediating receptors, inhibition of mediator synthesis, prevention of mediator release, anti-IgE therapy, corticosteroids, decreasing tumor burden, avoiding triggers, or a combination of these approaches [23, 48, 49]. Drugs used include H1-antihistamines (cetirizine, loratadine, or diphenhydramine), H2-antihistamines (famotidine or ranitidine), leukotriene inhibitors (montelukast, zafirlukast), MC stabilizers (cromolyn oral or inhalation, ketotifen available in some countries), corticosteroids (short-term use), epinephrine auto-injectors, and anti-IgE therapy (omalizumab). Dietary modifications with a low-histamine diet (like fermented foods, aged cheese, alcoholic beverages) and avoidance of triggers (physical or emotional stress, and extreme temperatures) and medications (e.g., opiates, NSAIDS, certain antibiotics, selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants (TCAs), angiotensin-converting enzyme (ACE) inhibitors, monoclonal antibodies, and so on) also could play an important role [26, 49, 50].
Emerging treatment options and active clinical trials
Brentuximab vedotin
In a phase II clinical trial, authors did not recommend brentuximab vedotin, an antibody drug conjugate, as monotherapy for patients with AdvSM [51].
Antibody drug conjugates are an interesting future perspective in MCL management.
Bezuclastinib
Apex trial studying bezuclastinib (CGT9486, a novel, highly selective, potent KIT D816V inhibitor) in AdvSM, including patients with MCL, is now actively recruiting patients [22, 52].
Early reports from this study showed that bezuclastinib was well tolerated with no serious grade 4 adverse events and there were notable reductions in disease markers like bone marrow MC burden, serum tryptase levels, and KIT D816V variant allele fractions [52].
Elenestinib
There is an active multicenter, open-label, two-arm study that is investigating elenestinib (BLU263, a different TKI that targets KIT D816V mutation) as monotherapy and with azacitidine (an antimetabolite), in AdvSM and other KIT-positive hematologic neoplasms [53, 54].
Tagraxofusp
Tagraxofusp (SL-401, an agent targeting IL-3 receptors) was initially developed for patients with blastic plasmacytoid dendritic cell neoplasms [55].
It is currently being studied in various hematological malignancies like AML, MDS, advanced hypereosinophilic disorder, and chronic myeloid leukemia (CML) [56]. Ongoing, multicenter, actively recruiting clinical trials are now studying tagraxofusp in patients with SM, including MCL [57, 58].
Ripretinib
Ripretinib (DCC-2618, an oral switch-control kinase inhibitor) was initially developed to treat gastrointestinal stromal tumors (GISTs).
There is an active clinical trial registered (but not yet recruiting patients) to study the tolerability, safety, and pharmacokinetics of ripretinib, in patients with advanced malignancies, including AdvSM [59].
Gemtuzumab ozogamicin (GO)
GO, a CD33-targeted drug conjugate, has been tested in some studies for patients with multimutated AdvSM, particularly refractory MCL and MCL-AHN (AML) [45]. So far, GO has been tried in isolated cases, and large-scale studies need to be conducted to assess its efficacy and safety profile [46, 60].
Other treatments
Immune checkpoint inhibitors (like anti-programmed cell death (PD)-1 or anti-cytotoxic T-lymphocyte associated protein (CTLA)-4 antibodies) have not been studied in MCL and are still being investigated in clinical trials for other hematologic malignancies. However, their potential role in MCL is an area of theoretical interest given their immunotherapeutic properties [61].
IL-2 has been used in basic experimental studies for some MC disorders to control symptoms and decrease MC burden, but its specific role in SM/MCL is not well-established, and the evidence supporting its efficacy is limited. Theoretically, it should help with cutaneous symptoms and lesions [6, 7].
To summarize, for patients with AdvSM including MCL, most studies suggest that either midostaurin or avapritinib should be considered as a first-line therapy rather than 2-CdA, other TKIs, IFN-α, or other agents, particularly if positive for KIT D816V mutation. Midostaurin and avapritinib have shown better response versus toxicity profile compared to other treatment regimens. We could not find any studies that have directly compared the efficacy and toxicity of these two drugs in MCL. Midostaurin is chosen over avapritinib if the platelet count is < 50 (× 103 cells/µL). For MCL patients with high MC burden, tumor debulking could be achieved with chemotherapeutic agents before using targeted therapies or HSCT. HSCT remains the mainstay of treatment in younger patients with compatible donors because of high rates of remission and survival. HSCT is also the main alternative in MCL patients who relapsed after chemotherapy or targeted therapy or have multi-mutated resistant disease (Fig. 13, treatment algorithm for MCL).
In conclusion, given the rarity and aggressive nature of MCL, treatment decisions should be individualized based on the disease characteristics, patient factors, and institutional expertise. Close collaboration with a multidisciplinary team, including hematologists, oncologists, and transplant specialists, is essential to optimize patient outcomes in this challenging disease.
| MCL Prognosis | ▴Top |
The main prognostic factor in SM is its subtype, with MCL having the worst prognosis. Older age (> 65 years), excessive weight loss, cytopenias, hypoalbuminemia, and percentage of bone marrow blasts (> 5%) are some of the poor prognostic factors observed in all types of SM, including MCL [8-10, 20, 22]. In addition, it is also observed that the patients with negative KIT D816V mutation have poor prognosis compared to patients with positive mutation, and the presence of S/A/R mutations indicates worse prognosis [8-10, 14, 20].
MCL has an OS ranging between 0 and 2.5 years, with a median OS for all types of MCL around 1.5 years. Acute MCL presents with severe organ damage and hence tends to be highly aggressive, carrying an extremely poor prognosis with a median OS of 2 - 3 months [8-10, 22, 25]. In multiple case reports on acute MCL, it is observed that the patients died within the first 2 months of diagnosis, irrespective of treatment [25, 62-66].
Going into the other subtypes of MCL, MCL-AHN has worse OS compared to MCL alone, and leukemic MCL has an inferior OS than the aleukemic form [8, 9, 14, 20]. No difference in survival rates was observed between primary and secondary MCL [8-10, 14, 20, 22].
| Conclusion | ▴Top |
MCL is a very rare and aggressive variant of SM, diagnosed when the SM criteria are met and when there are more than 20% atypical or immature MCs in the bone marrow biopsy. The most common molecular abnormality observed in MCL is mutations in the KIT gene, specifically the D816V mutation. Further research is essential to identify additional molecular abnormalities that could be targeted. Acute MCL with C-findings tends to be highly aggressive and has a very poor prognosis, with a median OS of 2 - 3 months. HSCT and newer TKIs targeting the KIT D816V pathway (mainly midostaurin and avapritinib, which are approved for SM) have shown promising outcomes in various types of MCL. Supportive measures to manage complications such as cytopenias, infections, TLS, MCAS, and organ failure also play an equally important role in managing MCL. Much more research and advancements are necessary before developing treatments that could further improve median OS.
Acknowledgments
None to declare.
Financial Disclosure
The authors declare that they do not have a financial relationship with any commercial entity that has an interest in the subject of this manuscript.
Conflict of Interest
The authors declare that they do not have any conflict of interest.
Author Contributions
All the authors actively participated in this review before submission. Dr. Idamakanti started the review, actively wrote different sections of the article, gathered images, conducted a literature review, and added references. Dr. Ebaid contributed to different sections of the article, actively edited and revised the article with current guidelines, conducted a literature review, and added references, as the attending hematologist. Dr. Bijjam contributed to different sections of the article and helped with proofreading (grammar) and revising the article. Dr. Bakhirev provided histopathology slides and input on pathogenesis and immunocytochemistry.
Data Availability
The authors declare that data supporting the findings of this review are available within the article.
Abbreviations
ACE: angiotensin-converting enzyme; AdvSM: advanced systemic mastocytosis; AIHA: autoimmune hemolytic anemia; AML: acute myeloid leukemia; ARDS: acute respiratory distress syndrome; ASM: aggressive systemic mastocytosis; ASXL: additional sex combs-like; BM: bone marrow; CBL: Casitas B-lineage lymphoma; CD: cluster differentiation; CM: cutaneous mastocytosis; CMML: chronic myelomonocytic leukemia; CNS: central nervous system; CT: computed tomography; CTLA: cytotoxic T-lymphocyte associated protein; ERK: extracellular signal-regulated kinase; FDA: Food and Drug Administration; GI: gastrointestinal; GvHD: graft-versus-host disease; HSCT: hematopoietic stem cell transplantation; IFN: interferon; IgE: immunoglobulin E; IL: interleukin; ILD: interstitial lung disease; ISM: indolent systemic mastocytosis; ITP: immune thrombocytopenia; JAK/STAT: Janus tyrosine kinase/signal transducers and activators of transcription; KIT: receptor tyrosine kinase type III; MC: mast cell; MCAS: mast cell activation syndrome; MCL: mast cell leukemia; MCS: mast cell sarcoma; MDS: myelodysplastic neoplasm; MPN: myeloproliferative neoplasm; MRI: magnetic resonance imaging; NHLBI: National Heart, Lung, and Blood Institute; NSAIDs: non-steroidal anti-inflammatory drugs; ORR: overall response rate; OS: overall survival; PCR: polymerase chain reaction; PD: programmed cell death; RAS: rat sarcoma; RFLP: restriction fragment length polymorphism; RUNX1: runt-related transcription factor 1; SM: systemic mastocytosis; SM-AHN: systemic mastocytosis-associated hematologic neoplasm; SRSF2: serine and arginine rich splicing factor 2; SSM: smoldering systemic mastocytosis; SSRI: selective serotonin reuptake inhibitor; TCAs: tricyclic antidepressants; TET: ten-eleven translocation; TLS: tumor lysis syndrome; WHO: World Health Organization
| References | ▴Top |
- Valent P, Akin C, Hartmann K, Alvarez-Twose I, Brockow K, Hermine O,
Niedoszytko M, et al. Updated diagnostic criteria and classification of mast cell disorders: a
consensus proposal. Hemasphere. 2021;5(11):e646.
doi pubmed - Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, Bejar
R, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid
Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36(7):1703-1719.
doi pubmed - Nahla AMH. The 2023 updated classification and diagnostic criteria of mastocytosis. Canc Therapy & Oncol Int J. 2023;24(4):556141.
- Wang SA, Orazi A, Gotlib J, Reiter A, Tzankov A, Hasserjian RP, Arber
DA, et al. The international consensus classification of eosinophilic disorders and systemic
mastocytosis. Am J Hematol. 2023;98(8):1286-1306.
doi pubmed - Pardanani A. Systemic mastocytosis in adults: 2021 Update on
diagnosis, risk stratification and management. Am J Hematol.
2021;96(4):508-525.
doi pubmed - Castells M, Metcalfe DD, Escribano L. Diagnosis and treatment of
cutaneous mastocytosis in children: practical recommendations. Am J Clin Dermatol.
2011;12(4):259-270.
doi pubmed - Matito A, Azana JM, Torrelo A, Alvarez-Twose I. Cutaneous
mastocytosis in adults and children: new classification and prognostic factors. Immunol Allergy
Clin North Am. 2018;38(3):351-363.
doi pubmed - Gilreath JA, Tchertanov L, Deininger MW. Novel approaches to treating
advanced systemic mastocytosis. Clin Pharmacol. 2019;11:77-92.
doi pubmed - Zanelli M, Quintini M, Magnasco S, Aprile L, Palicelli A, Zizzo M,
Sanguedolce F, et al. Mast cell leukemia: an update with a practical review. Cancers (Basel).
2023;15(6):1664.
doi pubmed - Georgin-Lavialle S, Lhermitte L, Dubreuil P, Chandesris MO, Hermine
O, Damaj G. Mast cell leukemia. Blood. 2013;121(8):1285-1295.
doi pubmed - Valent P, Sotlar K, Sperr WR, Reiter A, Arock M, Horny HP. Chronic
mast cell leukemia: a novel leukemia-variant with distinct morphological and clinical features.
Leuk Res. 2015;39(1):1-5.
doi pubmed - Lee HJ. Recent advances in diagnosis and therapy in systemic
mastocytosis. Blood Res. 2023;58(S1):96-108.
doi pubmed - Galli SJ, Dvorak AM, Dvorak HF. Basophils and mast cells: morphologic
insights into their biology, secretory patterns, and function. Prog Allergy.
1984;34:1-141.
pubmed - Kennedy VE, Perkins C, Reiter A, Jawhar M, Lubke J, Kluin-Nelemans
HC, Shomali W, et al. Mast cell leukemia: clinical and molecular features and survival outcomes
of patients in the ECNM Registry. Blood Adv. 2023;7(9):1713-1724.
doi pubmed - Arock M, Hoermann G, Sotlar K, Hermine O, Sperr WR, Hartmann K,
Brockow K, et al. Clinical impact and proposed application of molecular markers, genetic
variants, and cytogenetic analysis in mast cell neoplasms: Status 2022. J Allergy Clin
Immunol. 2022;149(6):1855-1865.
doi pubmed - Arock M, Sotlar K, Akin C, Broesby-Olsen S, Hoermann G, Escribano L,
Kristensen TK, et al. KIT mutation analysis in mast cell neoplasms: recommendations of the
European Competence Network on Mastocytosis. Leukemia. 2015;29(6):1223-1232.
doi pubmed - Schwaab J, Schnittger S, Sotlar K, Walz C, Fabarius A, Pfirrmann M,
Kohlmann A, et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood.
2013;122(14):2460-2466.
doi pubmed - Jawhar M, Schwaab J, Meggendorfer M, Naumann N, Horny HP, Sotlar K,
Haferlach T, et al. The clinical and molecular diversity of mast cell leukemia with or without
associated hematologic neoplasm. Haematologica. 2017;102(6):1035-1043.
doi pubmed - Pardanani AD, Lasho TL, Finke C, Zblewski DL, Abdelrahman RA, Wassie
EA, Gangat N, et al. ASXL1 and CBL mutations are independently predictive of inferior survival
in advanced systemic mastocytosis. Br J Haematol. 2016;175(3):534-536.
doi pubmed - Lim KH, Tefferi A, Lasho TL, Finke C, Patnaik M, Butterfield JH,
McClure RF, et al. Systemic mastocytosis in 342 consecutive adults: survival studies and
prognostic factors. Blood. 2009;113(23):5727-5736.
doi pubmed - Alvarez-Twose I, Matito A, Morgado JM, Sanchez-Munoz L, Jara-Acevedo
M, Garcia-Montero A, Mayado A, et al. Imatinib in systemic mastocytosis: a phase IV clinical
trial in patients lacking exon 17 KIT mutations and review of the literature. Oncotarget.
2017;8(40):68950-68963.
doi pubmed - Reiter A, George TI, Gotlib J. New developments in diagnosis,
prognostication, and treatment of advanced systemic mastocytosis. Blood.
2020;135(16):1365-1376.
doi pubmed - Rossini M, Zanotti R, Viapiana O, Tripi G, Orsolini G, Idolazzi L,
Bonadonna P, et al. Bone involvement and osteoporosis in mastocytosis. Immunol Allergy Clin
North Am. 2014;34(2):383-396.
doi pubmed - King JJ, Crawford EA, Iwenofu OH, Fox EJ. Case report: pathologic
long bone fracture in a patient with systemic mastocytosis. Clin Orthop Relat Res.
2007;459:263-269.
doi pubmed - Idamakanti M, Ebaid A, Bijjam RI, Bakhirev A, Mukkamalla SK,
Andritsos L. A rare case of acute aleukemic mast cell leukemia with osteoblastic lesions in the
appendicular skeleton. J Hematol. 2025;14(1):32-37.
doi pubmed - Akin C, Valent P, Metcalfe DD. Mast cell activation syndrome:
proposed diagnostic criteria. J Allergy Clin Immunol.
2010;126(6):1099-1104.e1094.
doi pubmed - Sciume M, De Magistris C, Galli N, Ferretti E, Milesi G, De Roberto
P, Fabris S, et al. Target therapies for systemic mastocytosis: an update. Pharmaceuticals
(Basel). 2022;15(6):738.
doi pubmed - Piris-Villaespesa M, Alvarez-Twose I. Systemic mastocytosis:
following the tyrosine kinase inhibition roadmap. Front Pharmacol. 2020;11:443.
doi pubmed - Ustun C, Keklik Karadag F, Linden MA, Valent P, Akin C. Systemic
mastocytosis: current status and challenges in 2024. Blood Adv. 2025;9(9):2048-2062.
doi pubmed - Gotlib J, Kluin-Nelemans HC, George TI, Akin C, Sotlar K, Hermine O,
Awan FT, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis.
N Engl J Med. 2016;374(26):2530-2541.
doi pubmed - Verstovsek S, Tefferi A, Cortes J, O'Brien S, Garcia-Manero G,
Pardanani A, Akin C, et al. Phase II study of dasatinib in Philadelphia chromosome-negative
acute and chronic myeloid diseases, including systemic mastocytosis. Clin Cancer Res.
2008;14(12):3906-3915.
doi pubmed - Zhu S, Jung J, Victor E, Arceo J, Gokhale S, Xie P. Clinical trials
of the BTK inhibitors ibrutinib and acalabrutinib in human diseases beyond B cell malignancies.
Front Oncol. 2021;11:737943.
doi pubmed - Ibrutinib in treating patients with advanced systemic mastocytosis. NCT02415608. September, 2018. Clinicaltrials.gov.
- DeAngelo DJ, Radia DH, George TI, Robinson WA, Quiery AT, Drummond
MW, Bose P, et al. Safety and efficacy of avapritinib in advanced systemic mastocytosis: the
phase 1 EXPLORER trial. Nat Med. 2021;27(12):2183-2191.
doi pubmed - Gotlib J, Reiter A, Radia D, Deininger M, George T, Panse J, et al.
P1023: avapritinib in patients with advanced systemic mastocytosis (ADVSM): efficacy and safety
analyses from the phase 2 pathfinder study with 2-year follow-up. Hemasphere.
2023;7(Suppl):e3184876.
doi - Gotlib J, Castells M, Elberink HO, Siebenhaar F, Hartmann K,
Broesby-Olsen S, George TI, et al. Avapritinib versus placebo in indolent systemic mastocytosis.
NEJM Evid. 2023;2(6):EVIDoa2200339.
doi pubmed - Valent P, Akin C, Sperr WR, Escribano L, Arock M, Horny HP, Bennett
JM, et al. Aggressive systemic mastocytosis and related mast cell disorders: current treatment
options and proposed response criteria. Leuk Res. 2003;27(7):635-641.
doi pubmed - Valent P, Sperr WR, Akin C. How I treat patients with advanced
systemic mastocytosis. Blood. 2010;116(26):5812-5817.
doi pubmed - Helbig G, Koclega A, Gawel WB, Wlodarczyk M, Rodzaj M, Labedz A, Hus
I, et al. The efficacy of cladribine (2-CdA) in advanced systemic mastocytosis.
Indian J Hematol Blood Transfus. 2020;36(4):661-666.
doi pubmed - Ustun C, Reiter A, Scott BL, Nakamura R, Damaj G, Kreil S, Shanley R,
et al. Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J Clin
Oncol. 2014;32(29):3264-3274.
doi pubmed - Hauswirth AW, Simonitsch-Klupp I, Uffmann M, Koller E, Sperr WR,
Lechner K, Valent P. Response to therapy with interferon alpha-2b and prednisolone in aggressive
systemic mastocytosis: report of five cases and review of the literature. Leuk Res.
2004;28(3):249-257.
doi pubmed - Cladribine plus pegylated interpheron Alfa-2a in systemic mastocytosis. NCT01602939. 2016. Clinicaltrials.gov.
- Gruson B, Lortholary O, Canioni D, Chandesris O, Lanternier F,
Bruneau J, Grosbois B, et al. Thalidomide in systemic mastocytosis: results from an open-label,
multicentre, phase II study. Br J Haematol. 2013;161(3):434-442.
doi pubmed - Treatment of indolent systemic mastocytosis with PA101. NCT02478957. 2016. Clinicaltrials.gov.
- Bauer J, Longo W, Yang D. Mast cell leukemia: review of a rare disease and case report of prolonged survival after allogeneic stem cell transplant. Hum Pathol Case Rep. 2017;10:46-49.
- Valent P, Akin C, Arock M, Gleixner KV, Greinix H, Hermine O, Horny
HP, et al. Antibody-based and cell therapies for advanced mastocytosis: established and novel
concepts. Int J Mol Sci. 2023;24(20):15125.
doi pubmed - Spyridonidis A, Thomas AK, Bertz H, Zeiser R, Schmitt-Graff A,
Lindemann A, Waller CF, et al. Evidence for a graft-versus-mast-cell effect after allogeneic
bone marrow transplantation. Bone Marrow Transplant. 2004;34(6):515-519.
doi pubmed - Stem cell transplantation to treat systemic mastocytosis. NCT00006413. 2017. Clinicaltrials.gov.
- Castells M, Butterfield J. Mast cell activation syndrome and
mastocytosis: initial treatment options and long-term management. J Allergy Clin Immunol
Pract. 2019;7(4):1097-1106.
doi pubmed - Matito A, Escribese MM, Longo N, Mayorga C, Luengo-Sanchez O,
Perez-Gordo M, Matheu V, et al. Clinical approach to mast cell activation syndrome: a practical
overview. J Investig Allergol Clin Immunol. 2021;31(6):461-470.
doi pubmed - Gotlib J, Baird JH, George TI, Langford C, Reyes I, Abuel J, Perkins
C, et al. A phase 2 study of brentuximab vedotin in patients with CD30-positive advanced
systemic mastocytosis. Blood Adv. 2019;3(15):2264-2271.
doi pubmed - DeAngelo DJ, Pullarkat V, Piris-Villaespesa M, George TI, Patel JL,
Ustun C, et al. P1049: a phase 2 study of Bezuclastinib (CGT9486), a novel, highly selective,
potent KIT D816V inhibitor, in adults with advanced systemic mastocytosis (APEX): METHODS,
baseline data, and early insights. Hemasphere. 2022;6(Suppl):939-940.
doi - A Phase 1/2 Study of Elenestinib (BLU-263) as Monotherapy and in Combination With Azacitidine in Patients With Advanced Systemic Mastocytosis (AdvSM) and and Other KIT Altered Hematologic Malignancies. NCT05609942. April, 2024. Clinicaltrials.gov.
- DeAngelo DJ, Reiter A, George TI, et al. AZURE: a phase 1/2 study of
blu-263 as monotherapy and in combination with azacitidine in patients with advanced systemic
mastocytosis. Blood (ASH Annual Meeting Abstracts). 2022;140(Suppl 1):6877-6978.
doi - Frankel AE, Woo JH, Ahn C, Pemmaraju N, Medeiros BC, Carraway HE,
Frankfurt O, et al. Activity of SL-401, a targeted therapy directed to interleukin-3 receptor,
in blastic plasmacytoid dendritic cell neoplasm patients. Blood. 2014;124(3):385-392.
doi pubmed - SL-401 in combination with azacitidine or azacitidine/venetoclax in acute myeloid leukemia (AML), high-risk myelodysplastic syndrome (MDS) or blastic plasmacytoid dendritic cell neoplasm (BPDCN). 2017. Updated November 2, 2021. Accessed April 21, 2022. Cinicaltrials.gov.
- SL-401 in advanced, high risk myeloproliferative neoplasms (systemic mastocytosis, advanced symptomatic hypereosinoophic disorder, chronic myelomonocytic leukemia). Mayo Clinic. Accessed April 21, 2022. Clinicaltrials.gov.
- Patnaik MM, Gupta V, Gotlib JR, Carraway HE, Wadleigh M, Schiller GJ, et al. Results from ongoing phase 2 trial of SL-401 in patients with advanced, high-risk myeloproliferative neoplasms including chronic myelomonocytic leukemia, systemic mastocytosis, advanced symptomatic hypereosinoophic disorder. Blood. 2016;128(22):4245.
- A safety, tolerability, and PK study of DCC-2618 in patients with advanced malignancies. 2015. Updated April 20, 2021. Accessed April 21, 2022. Clinicaltrials.gov.
- Alvarez-Twose I, Martinez-Barranco P, Gotlib J, Garcia-Montero A,
Morgado JM, Jara-Acevedo M, Merker JD, et al. Complete response to gemtuzumab ozogamicin in a
patient with refractory mast cell leukemia. Leukemia. 2016;30(8):1753-1756.
doi pubmed - Ribatti D. New insights into the role of mast cells as a therapeutic
target in cancer through the blockade of immune checkpoint inhibitors. Front Med (Lausanne).
2024;11:1373230.
doi pubmed - Ender E, Dages K, Pitlick M, Voelker D, Pongdee T. Mast cell leukemia: a case report. Annals of Allergy, Asthma & Immunology. 2022;129(5):S164.
- Galura GM, Cherukuri SV, Hakim N, Gaur S, Orazi A. Acute aleukemic
mast cell leukemia: Report of a case and review of the literature. Leuk Res Rep.
2020;14:100230.
doi pubmed - Zeerleder S, van Oers M. Aleukemic variant of mast cell leukemia.
Blood. 2012;119(9):1961.
doi pubmed - Vasilj A, Katovic SK, Maricevic I, Zokvic E, Gacina P. [Mast cell
leukemia—case report]. Acta Med Croatica. 2013;67(1):61-64.
pubmed - Jafari E, Hadipour A, Kalantari Khandani B, Abolhasani F. Mast cell
leukemia with ascites and multiple organs damage. Iran J Pathol.
2019;14(3):265-269.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Hematology is published by Elmer Press Inc.