| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://jh.elmerpub.com |
Case Report
Volume 14, Number 5, October 2025, pages 273-280

Fever: Harmless Heat or a Deadly Inferno? Unmasking Hemophagocytic Lymphohistiocytosis and Beyond: A Rare Case of Mast Cell Sarcoma and Overview of HLH
Karan J. Yagnika, c ![]() , Adelaine Espiritua, Malay
Rathoda, Jillianne Unasa, Raymart Macasaeta, FNU
Payala, Patricia Perez De Taglea, Shazia Shaha, Priya
Angia, b, c
, Adelaine Espiritua, Malay
Rathoda, Jillianne Unasa, Raymart Macasaeta, FNU
Payala, Patricia Perez De Taglea, Shazia Shaha, Priya
Angia, b, c
aDepartment of Internal Medicine, Rutgers Health/Monmouth Medical Center, Long Branch,
NJ 07740, USA
bDepartment of Geriatrics, Rutgers Health/Monmouth Medical Center,
Long Branch, NJ 07740, USA
cCorresponding Authors: Karan J. Yagnik and Priya
Angi, Department of Internal Medicine, Monmouth Medical Center, Long Branch, NJ 07740, USA
Manuscript submitted May 24, 2025, accepted August 21, 2025, published online October 10,
2025
Short title: A Rare Case of MCS Presenting as HLH
doi:
https://doi.org/10.14740/jh2087
| Abstract | ▴Top |
Hemophagocytic lymphohistiocytosis (HLH) is a rare and potentially fatal hyperinflammatory syndrome characterized by uncontrolled activation of the immune system, often secondary to infections, autoimmune diseases, or malignancies. While malignancy-associated HLH is increasingly recognized, its diagnosis remains challenging due to nonspecific clinical features that mimic other systemic illnesses. Mast cell sarcoma (MCS), an exceedingly rare and aggressive neoplasm of aberrant mast cells, is an exceptionally uncommon cause of HLH and is rarely reported in the literature. We described the case of a 70-year-old male who presented with persistent fever, weight loss, night sweats, and pancytopenia. Initial investigations for infectious, autoimmune, and neoplastic etiologies were inconclusive. The patient rapidly deteriorated despite empirical antimicrobial therapy and supportive care. Serial imaging revealed hepatosplenomegaly, and laboratory studies showed markedly elevated ferritin, lactate dehydrogenase (LDH), and soluble interleukin (IL)-2 receptor levels. Bone marrow biopsy demonstrated hemophagocytosis, and he fulfilled both HLH-2004 criteria and had a high HScore, confirming the diagnosis of HLH. Despite initiating HLH-directed therapy with high-dose corticosteroids and etoposide, the patient’s condition progressed rapidly, leading to multiorgan failure and death. Post-mortem examination revealed extensive infiltration of atypical mast cells in the bone marrow, spleen, liver, and lymph nodes. Immunohistochemistry confirmed the diagnosis of systemic MCS. This report highlights the diagnostic complexity of HLH, particularly when triggered by rare malignancies like MCS. The case emphasizes the need for high index of suspicion and early, aggressive diagnostic workup - including bone marrow evaluation and molecular studies - in patients presenting with fever of unknown origin, cytopenias, and systemic inflammation. Early identification of malignancy-associated HLH is critical, as targeted treatment of the underlying disease is often necessary for a favorable outcome. Awareness of atypical presentations and rare triggers is essential for timely diagnosis and management of this life-threatening syndrome.
Keywords: Hemophagocytic lymphohistiocytosis; Fever of unknown origin; Mast cell sarcoma; HLH-2004; HScore
| Introduction | ▴Top |
Hemophagocytic lymphohistiocytosis (HLH) is a rare and fatal condition with a high mortality rate of 20-80% [1-7]. Mortality rate of secondary HLH (sHLH) depends upon underlying triggers such as malignancy, infection, drugs or autoimmune condition. Malignancy is the most common cause of sHLH [8]. HLH due to malignancy typically has a higher mortality rate than non-malignant triggers [1, 9, 10]. Parikh et al have shown that the median survival rate in malignancy associated HLH is 1.4 months in contrast to 22.8 months for non-malignancy associated HLH [5]. Malignant triggers of HLH are usually hematological neoplasms such as T-cell, natural killer (NK)-cell, B-cell lymphomas, Hodgkin’s lymphoma and leukemias, especially acute myeloid leukemia (AML) undergoing chemotherapy [11]. However, uncommonly solid tumors can also be associated with HLH.
Mast cell sarcoma (MCS) is an exceedingly rare and aggressive type of cancer that originates from mast cells and characterized by abnormal accumulation of atypical mast cells in one or more organs [12]. Prognosis is very poor with a median survival rate of 18 months and often results as fatal due to failure of conventional chemotherapies [13]. Progression to mast cell leukemia is also not unusual. Exact underlying pathophysiology is unclear; however, it is believed that mutation of the KIT gene is involved. Diagnosis is done by biopsy of tumors and immunostaining of mast cells. However, clinical presentation can be variable and often results in delayed diagnosis due to resembling multiple differential diagnosis [14]. Hence, very limited data are available on such tumors. Surgery, radiation, chemotherapy or targeted therapy can be useful; however, due to high failure rates, exact guidelines of therapy are still under investigation with combination of therapies such as targeted therapies to KIT gene, 2-CdA, anti-CD30 and bone-marrow transplantation.
Here, we are adding a case in limited literature, which presented as fever of unknown origin (FUO). While the investigative workup ensued and HLH was diagnosed, the patient’s clinical condition deteriorated. Despite robust investigations and therapy, the patient eventually died. Afterwards, a post-mortem biopsy specimen resulted as MCS, and simultaneously pre-mortem bone marrow biopsy results came back and showed same diagnosis. This autopsy confirmation highlights the diagnostic challenges and clinical complexities of the case.
| Case Report | ▴Top |
A man in his 70s was sent to the hospital emergency room (ER) from his primary care doctor’s office due to a high-grade fever (105 °F). The patient reported daily fevers for the past 1 month, with a range of maximum temperature (Tmax) 104 - 105 °F, followed by profuse sweating. He also reported malaise, anorexia, fatigue, petechial rashes on abdomen and back and weight loss of 15 lb over the period of the past 1 month.
The patient had a significant past medical history of chronic thrombocytopenia and alcohol use disorder. Review of records revealed a prior hematology evaluation and thrombocytopenia with platelet count of 100,000/µL 5 years ago. According to the hematologist, the patient’s thrombocytopenia was attributed to fatty liver, hypersplenism and chronic alcohol intake after ruling out other potential causes. Five years later, he saw a new hematologist-oncologist physician after another referral from his primary care physician for down trending chronic thrombocytopenia and circulating myelocytes and metamyelocytes. Abdominal ultrasound revealed enlarged spleen (15.4 cm) and liver (15 cm) compared to prior ultrasound. At this time, complete blood count (CBC) revealed white blood cell (WBC) count of 5,320/µL, absolute neutrophil count 2,990/µL, hemoglobin 14.8 g/dL and platelet counts were 75,000/µL, and comprehensive metabolic profile (CMP) was within the normal limit. Vitamin B12 was high at 2, 205 pg/mL and folic acid was high at > 40 ng/mL. IntelliGEN Myeloid next-generation sequencing (NGS) assay was also done, demonstrating three Tier 1 and one Tier 2 abnormality, which is concerning for myelodysplastic syndrome (MDS). However, his new hematologist-oncologist suspected a causal relationship of this abnormality with alcohol ingestion and chronicity of the thrombocytopenia without leukopenia or anemia. Additionally, the early release of myeloid forms and the low level of myelocytes and metamyelocytes on earlier CBCs were thought to be attributed to chronic alcohol ingestion. However, he was requested to do iron profile, ferritin, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), JAK2 gene mutation with reflex to calretinin and MPL gene mutation, antiplatelet antibody profile, FibroScan, flow cytometry, bone marrow aspiration/biopsy, but the patient refused. Thus, the patient was only observed and advised to limit his alcohol intake.
Upon arrival at the ER, he was fully awake, alert, and oriented to person, place, time, and situation, without any acute distress. He was afebrile, sinus tachycardia on telemetry with heart rate (HR) in low 100s, BP 130/82 mm Hg, and respiratory rate (RR) 18/min on arrival. Physical examination showed grade 2/6 systolic murmur, hepatosplenomegaly, petechial rash on patient’s back and abdomen, otherwise grossly unremarkable. Electrocardiogram (EKG) showed sinus tachycardia with HR 125 beats per minute (bpm), QTc 447 ms, without any ST or T wave changes. Laboratory tests show sodium 132 mmol/L, total bilirubin 1.7 mg/dL, direct 0.8 mg/dL, WBC 8,000 cells/µL, platelets 39,000 cells/µL, neutrophil count 7,900 cells/µL, international normalized ratio (INR) 1.6. Point of care tests for influenza A and B as well as coronavirus disease 2019 (COVID-19) were negative. Chest X-ray was negative for any cardiopulmonary disease.
During the hospital course, the patient was spiking high-grade fevers daily (Tmax 105 - 106 °F). A full infectious workup was performed, including Lyme disease, Babesia, a tick-borne panel, parasitic panel, and dengue fever. Blood cultures demonstrated no growth throughout the hospital course, including confirmation with repeat cultures. Echo demonstrated normal ejection fraction (EF, 60-65%) and no valvular vegetations. Computed tomography (CT) chest abdomen pelvis with intravenous (IV) contrast showed hepatosplenomegaly but was otherwise unremarkable. Autoimmune panels were also unremarkable. He was empirically treated for 7 days with doxycycline without improvement.
On day 3, ferritin was elevated to > 4,000 ng/mL, and given his cytopenias, as well as hepatosplenomegaly on imaging and recurrent high fevers, there was concern for HLH. IL-2 receptor (CD25) testing was sent, which came back elevated at 32,210 ng/mL, further supporting the diagnosis of HLH. Liver biopsy was recommended but was unable to be performed due to thrombocytopenia. A bone marrow biopsy was performed on day 5 with flow cytometry and fluorescence in situ hybridization (FISH) MDS panel, which later demonstrated by hospital day 11 atypical cells expressing CD4, CD117, CD43, CD71, CD30, dim CD25, weak E-cadherin, while lacking expression of CD45, CD34, HLA-DR, s/cCD3, CD2, CD5, CD7, CD20, CD61, GPHA, CD56, CD57, TCR a/b, TCR g/d, tryptase, and CD123. It also showed hypercellular bone marrow with maturing trilineage hematopoiesis, erythroid dyspoiesis (megaloblastoid maturation, binucleation, cytoplasmic blebs and vacuolization) and moderate reticulin fibrosis (MF-2), as well as increased histiocytes with occasional hematophagocytosis. It was also noted to have atypical large mononuclear cells, myelofibrosis, discohesive erythroid precursors, occasional dysmegakaryopoiesis (mononuclear with focal atypia) and increased reticuloendothelial system (RES) iron without ring sideroblasts. However, granulopoiesis and lymphocytes were unremarkable. Also, immunostaining patterns ruled out lymphoproliferative or plasma cell neoplasm. Given the expression of CD43 and other hematopoietic related markers, with a background of dysplastic erythroid precursors and megakaryocytes, decision was made that it could be suggestive of a high-grade myelodysplastic neoplasm, though further lineage could not be assigned. Furthermore, FISH and chromosomal analysis were also negative for any cytogenic alterations.
On hospital day 4, the patient developed respiratory distress. Oliguria with rising creatinine led to concern for volume overload, and the patient could not tolerate noninvasive ventilation due to hypotension. Hence, he eventually required intubation and pressor support. Later, he had a generalized seizure for which he was started on antiepileptic. Given continued concern for HLH, he was started on HLH protocol with etoposide twice weekly and dexamethasone daily. He was also started on empiric meropenem to cover possible sepsis and was subsequently transferred to tertiary center for higher level of care on the following day.
At the tertiary care hospital, the calculated HScore continued to be elevated at 224, and he was also found to have findings consistent with tumor lysis syndrome, including electrolyte abnormalities and markedly elevated uric acid level (14.8 mg/dL), for which rasburicase and allopurinol were started. Video electroencephalogram (EEG) showed findings consistent with toxic metabolic encephalopathy. Over the few days after starting HLH protocol with etoposide and dexamethasone, though inflammatory markers were slightly improving, fever was replaced by an episode of hypothermia, for which pulse dose methylprednisolone 1 g was initiated for 3 days. He was started on combination chemotherapy with etoposide/cytarabine three times per week, as well as a tapered dexamethasone regimen per protocol. Prophylactic antimicrobials with acyclovir, Bactrim, and micafungin three times per week for immunocompromised state were also initiated.
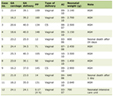
On hospital day 11, the patient’s code status was changed to partial code - not to resuscitate but okay for intubation. On the subsequent days, the patient was able to be weaned off sedation and eventually pressors, and the patient was noted to have an improving mental status although remained lethargic. Later, he was started on intravenous immunoglobulin (IVIG) for refractory thrombocytopenia. Although the patient improved briefly for a few days, his condition reversed and he became more lethargic, febrile, hypotensive requiring pressor support, and with severe neutropenia. CT scan of the abdomen and pelvis this time showed new interstitial pancreatitis. Given suspicion for neutropenic sepsis secondary to chemotherapy, empiric vancomycin and meropenem were started. Dexamethasone taper was also discontinued. Throughout this hospital course, multiple hematological, pathological and molecular testing were performed, none of which showed clear evidence of underlying etiology for the HLH. Biopsy samples were also sent to the National Institutes of Health (NIH) for further study, which, after patient’s demise, came back later as myeloid neoplasm consistent with MCS, involving hypercellular marrow (about 33% atypical cells, 95% cellularity with mild dyserythropoiesis and dysmegakaryopoiesis and associated systemic mastocytosis and hemophagocytosis, CD117-positive malignant population, 29% on flow cytometry and myeloid genetic panel with KIT variant with amino acid change p.Asp816Val and other myeloid disorder related mutations). Immunophenotyping showed positivity for CD117, CD71, CD4, CD43, CD30, dim E-cadherin, and dim CD25 and negativity for CD45, ALK, (MF-1+) CD3, CD2, CD5, CD7, CD8, CD20, PAX5, CD68, tryptase, OSCAR, CAM5.2 CK8/18, OCT4, glypican, SOX10, PLAP, ALK1, CD68, lysozyme CD138, CD34, CD61, MPO, glycophorin, hemoglobin A, and EBER-ISH. Also, CD34 does not show an increase in myeloblasts. Bone marrow biopsy (from posterior iliac crest) and autopsy confirmed involvement of bone marrow, spleen and lymph nodes. Genetic analysis results are shown in Table 1.
 Click to view |
Table 1. Genetic Analysis Results |
On the hospital day 17, due to clinical deterioration despite extreme measures, he was given comfort care by his family, and terminal extubation was done. Autopsy showed findings consistent with HLH secondary to MCS. Full hospital course is summarized in Figure 1.
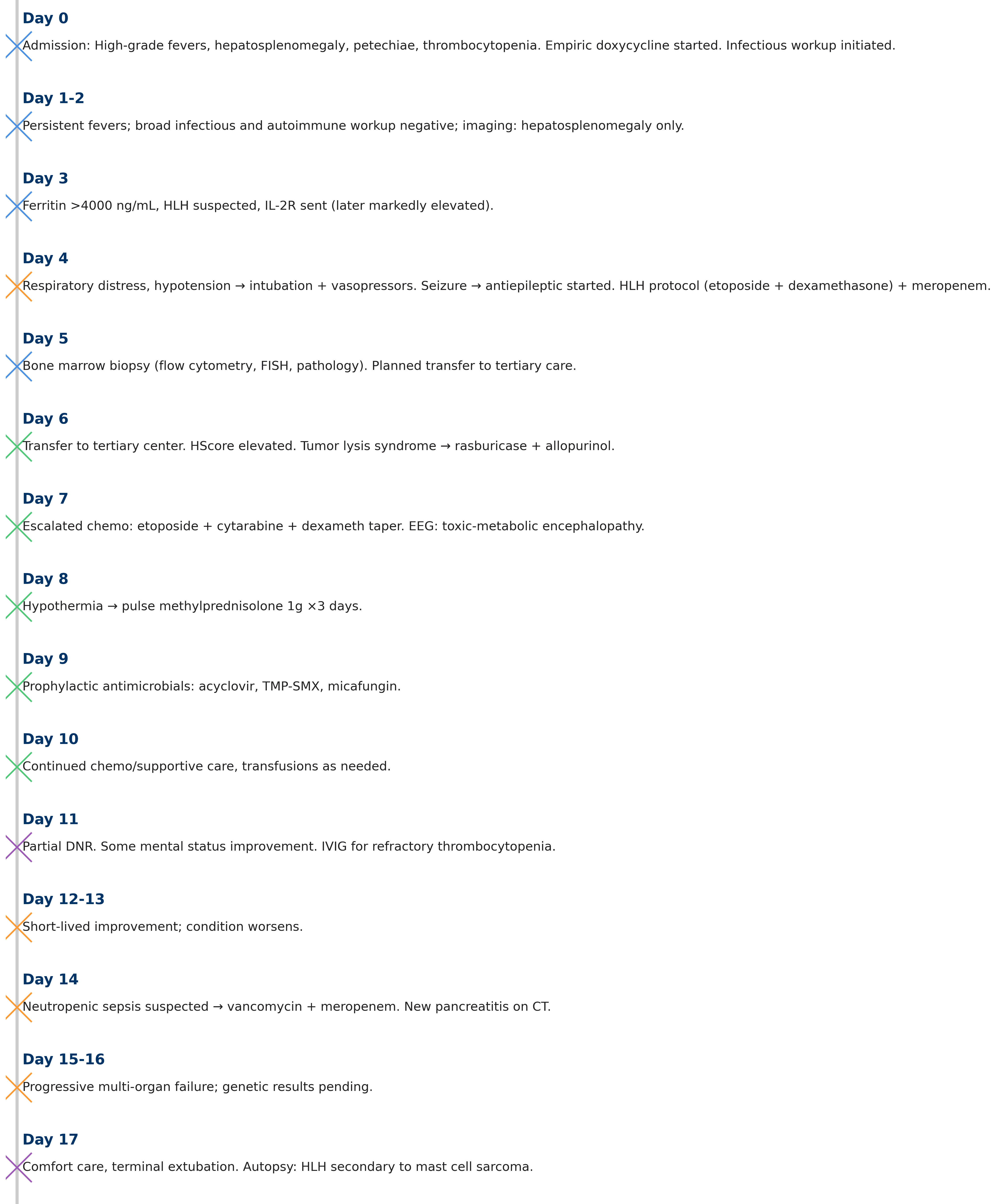 Click for large image |
Figure 1. Day-by-day hospital course with corresponding management. HLH: hemophagocytic lymphohistiocytosis; IL-2R: interleukin 2 receptor; EEG: electroencephalogram; FISH: fluorescence in situ hybridization; IVIG: intravenous immunoglobulin; CT: computed tomography; DNR: do-not-resuscitate. |
| Discussion | ▴Top |
We report a unique case of MCS, presented as FUO, manifested as HLH. Dr. Petersdorf and Dr Beeson first proposed the definition of FUO in 1961. They defined FUO as a temperature above 101 °F (38.3 °C) for more than 3 weeks with no diagnosis after at least a week of hospital evaluation [15]. As practice has evolved, with more advanced outpatient investigations, more immunocompromised patients and more complex therapeutic interventions, the original definition has been modified accordingly [16].
Detailed clinical history and physical examination are crucial first steps in FUO assessment and will direct further investigations. A recommended diagnostic workup includes a CBC with differential, three sets of blood cultures from various sites at varying times before any antibiotic therapy is administered, chest radiography, complete metabolic panel (with hepatitis serologies if liver function tests are abnormal), urinalysis with microscopy and culture, ESR, CRP, antinuclear antibody (ANA), rheumatoid factor (RF), cytomegalovirus (CMV) immunoglobulin (Ig)M or viral DNA testing, heterophile antibody test, tuberculin skin testing, human immunodeficiency virus (HIV) screening, and abdominal CT scan [17]. In our case, appropriate initial diagnostic workup was performed, and appropriate subspecialists were consulted as well.
Our patient presented to the ER with a complaint of fever for at least 1 month, along with other symptoms including malaise, fatigue, rash, anorexia and weight loss, with persistent low platelet counts, prompting a more robust investigation for malignancy. Differential diagnoses included lymphomas, renal cell carcinoma, acute leukemias like AML, myeloproliferative disorders, and breast, liver, pancreas, or colon cancer, in addition to less frequent entities like malignant histiocytosis [18].
This case represents a complex interplay between diagnostic challenges for HLH, and its management. HLH was initially described by Farquhar and Claireaux in 1952. HLH is a potentially fatal inflammatory condition that is marked by the uncontrollable activation of histiocytes and macrophages, leading to the phagocytosis of blood cells within the bone marrow [19]. There are two clinical types of HLH. Primary HLH and sHLH. Primary HLH is more common in children. sHLH is present in adults with a mean age of 50 years. Classical triggers of sHLH are autoimmune diseases, infections, metabolic disorders and malignancy. In adults from North America and Europe, HLH more often manifests as a secondary condition. Among which, half are due to cancers, with hematologic malignancies being notable. Primary or inherited HLH is uncommon in adults [20].
Clinical features of HLH include prolonged fever, anemia, thrombocytopenia and leukopenia, hepatosplenomegaly, elevated liver enzymes, elevated ferritin, elevated triglycerides, fatigue and malaise. HLH secondary to malignancy (mHLH) is especially challenging to diagnose, as many of its defining laboratory characteristics may potentially overlap with those commonly seen in malignancy itself. For instance, cytopenias and splenomegaly may be due to direct infiltration by leukemia or lymphoma; fever may be due to opportunistic infections in immunocompromised hosts or due to neoplastic processes; low fibrinogen may be due to disseminated intravascular coagulation (DIC) as a complication of malignancy; and elevated ferritin levels may be due to chronic inflammation or iron overload as a consequence of repeated transfusions. In this case, initially thrombocytopenia was thought to be attributed to chronic alcohol consumption, fatty liver and hypersplenism. Because of the insidious and overlapping presentation of HLH in adults, especially in the context of malignancy, a high index of suspicion is crucial. Special vigilance is needed in patients with unexplained fever, high ferritin, and bi- or pancytopenia. Because delays in diagnosis are common because of the time it takes to obtain specialized tests, early clinical suspicion and initiation of treatment are required to prevent sudden clinical decompensation and organ failure. Due to its broad range of triggers and typically nonspecific presentation, HLH can be difficult to diagnose. The diagnosis is either confirmed by identifying a disease-causing genetic mutation or by fulfilling five or more of a set of eight diagnostic findings.
The diagnosis of HLH can be established if either of the following criteria is fulfilled: 1) A molecular diagnosis consistent with HLH; or 2) Any five of the eight following clinical and laboratory criteria for HLH: a) fever > 38.5 °C; b) splenomegaly; c) cytopenia (affecting ≥ 2 of 3 lineages in peripheral blood: hemoglobin (Hb) < 9 g/dL (in infants < 4 weeks: Hb < 100 g/L), platelets < 100 × 109/L, neutrophils < 1.0 × 109/L); d) hypertriglyceridemia and/or hypofibrinogenemia: fasting triglycerides > 3.0 mmol/L (> 265 mg/dL) or fibrinogen ≤ 1.5 g/L; e) hemophagocytosis in bone marrow, spleen, liver, lymph nodes, or other tissues; f) low or absent NK-cell activity; g) serum ferritin concentration ≥500 µg/L; h) soluble CD25 (soluble IL-2 receptor) ≥ 2,400 U/mL [21].
To improve early diagnosis of HLH, a simplified diagnostic approach derived from the HLH-2004 criteria has been proposed. The abbreviated approach proposes to diagnose HLH in the presence of at least three of the following four clinical features: fever of a persistent nature, splenomegaly, cytopenias, and hepatitis. Additionally, one or more immunologic abnormalities should be identified, which may be increased ferritin, elevated levels of soluble interleukin-2 receptor alpha (sIL2R), very low or absent NK cell activity, or hemophagocytosis on tissue biopsy findings. To also aid diagnosis, the HScore was developed as a clinical tool to estimate the probability of HLH in adults. Based on weighted clinical and laboratory parameters, an HScore threshold of 169 was determined to be optimal, with 93% sensitivity and 86% specificity [20].
In a retrospective review at MD Anderson Cancer Center, only 21% of patients with suspected HLH met the initial HLH-2004 criteria. This led to demands for an expanded diagnosis system centered on 18 laboratory and clinical parameters, with the presence of any five being considered sufficient to establish a diagnosis [22]. Also, in a retrospective multicenter study involving 225 patients with hematologic malignancies, it was demonstrated that sIL2R > 3,601 U/mL and ferritin > 920 ng/mL were useful predictors of HLH with 88% sensitivity and 76% specificity [23].
Additionally, diagnosis of the underlying etiology of HLH is critical to guide both diagnosis and treatment. In malignancy-associated HLH (mHLH), diagnosis can be particularly problematic. The early course of HLH is often dominated by a profound cytokine storm, during which the underlying malignancy may not be apparent. The inability to make a tissue diagnosis can hinder the timely initiation of cancer-specific treatments. Additionally, mHLH is usually triggered by a trio of occurrences - malignancy, infection, and immune dysregulation - which demands stepwise and multidisciplinary treatment.
The clinical findings of HLH are also consistent among a literature review involving 23 cases of MCS, wherein mast cell activation symptoms were commonly observed - including flushing, fever, malaise, diarrhea, and tachycardia [13]. Most common organ involved is the bone (78%), with masses found in the thoracic vertebrae, pelvis, tibia, femur, ankle and skull; followed by the gastrointestinal tract (35%); lymph nodes (30%); skin (30%); spleen (26%); and liver (22%). MCS, a rare and aggressive form of cancer composed of highly atypical mast cells, was found to be the trigger of HLH in our case patient [24]. The markedly atypical morphology of these neoplastic cells frequently complicates diagnosis, owing to their resemblance to carcinoma metastases, anaplastic lymphomas, and histiocytic neoplasms [25, 26]. The cause of MCS is unknown [27]. Mast cells are a type of WBC of myeloid lineage that normally resides in tissues throughout the body and play a role in both normal functions and disease [28]. MCS typically shows staining positive for tryptase and/or CD117 [29]. Tryptase is mast cell-specific protease, and its presence is a key indicator of mast cell origin in sarcomas. However, tumor cells in MCS are highly atypical forms of mast cells, and sometimes it shows negative or weakly staining of tryptase even though it is a mast cell neoplasm [29, 30]. In fact, tryptase expression can be low or missing in 1-5% of cases of mastocytosis, resulting in CD117-only immunophenotype of mast cells and possibly delaying diagnosis, as seen in our case [29]. In addition to CD117, CD25 has been found to be more reliable marker as it does not express by normal/reactive mast cell but the transformed ones [29]. Moreover, mast cells may express a great variety of myeloid (MPO, 2D7, CD203c, naphthol AS-D chloroacetate esterase), monohistiocytic (CD14, CD68), stem cell-related (CD44), and lymphoid (CD2, CD30, CD52, CD79a) antigens in varying frequencies and intensities. Such immunophenotypic features of transformed mast cells may lead to severe diagnostic problems for the hematopathologist, as depicted in our case [29]. Limited literature demonstrates an abysmal prognosis [26]. With less than 30 cases of MCS reported in literature [31], the development of HLH manifesting as FUO from MCS represents a distinctive and infrequent clinical trajectory within an already uncommon disease entity. This thus poses a unique diagnostic challenge, particularly within the context of its underlying rarity.
Furthermore, in a minority of cases of MCS, patients who have had pre-existing or concurrent systemic mastocytosis are associated with a certain genetic mutation - a point mutation at codon 816 of the KIT proto-oncogene (abbreviated p.D816V) [27, 28]. The p.D816V mutation has only been reported in three cases of MCS, though there are an additional six reported cases of MCS with other KIT mutations. In the current case, the decedent was known to have chronic thrombocytopenia but did not have a known history of mastocytosis. A myeloid gene sequencing panel (which was performed months prior to the decedent’s presentation with HLH as part of his thrombocytopenia workup) detected the p.D816V mutation in the decedent’s blood sample. Also, other genetic mutations were detected that are associated with myelodysplastic disorders but whose relevance to the case are less clear.
To our knowledge, there is only one case report in which hemophagocytosis was noted in a patient with MCS. In this report, the patient died 2 months following diagnosis, and it is unknown whether the patient was diagnosed with HLH [32]. Treatment options for adult HLH are largely extrapolated from pediatric clinical trials. Previously, before the introduction of etoposide-based therapy, 5-year survival had been dismal, approximately 20% [33]. Literature suggests starting the management of the underlying malignancy at the earliest. The HLH-94 protocol, introduced by the Histiocyte Society, dramatically altered the outcome by using a combination of etoposide and corticosteroids to manage inflammation and treat the underlying etiology [34]. Over the past decade, there have been few changes in treatment. The HLH-2004 regimen included the addition of cyclosporine to the HLH-94 regimen; however, long-term survival is still 50% to 60% [21].
The role of HLH-94 in adult mHLH is contentious. One of the problems is the lack of prospective trials to guide treatment in this population. Moreover, upfront therapy with immunosuppressive agents such as dexamethasone and etoposide may interfere with curative cancer therapy. Despite these challenges, clinical practice guidelines mostly recommend that therapy for the underlying malignancy should be started as soon as possible. Corticosteroids are also often started at the same time to help control the hyperinflammatory process. If further immunosuppression is required but etoposide is contraindicated, biologic agents against IL-1 or IL-6 have been used.
One of the most promising experimental therapies is ruxolitinib, a JAK inhibitor with primary activity against JAK1 and JAK2 and secondary activity against TYK2 and JAK3. Since many cytokines involved in HLH signal through the JAK-STAT pathway, pathway inhibition has proven effective in dampening immune overactivation. Preliminary data also suggest that ruxolitinib can restore steroid sensitivity of CD8+ T cells. Additional investigational agents under investigation in early-phase trials in adult mHLH include venetoclax (NCT05546060), a BCL-2 inhibitor, and zanubrutinib (NCT05320575), a Bruton’s tyrosine kinase (BTK) inhibitor - both of which have demonstrated effectiveness in hematologic malignancies and may offer additional therapeutic benefit in HLH [20].
MCS management usually involves combination of therapy such as surgery, radiation, chemotherapy and target therapy depending upon extent disease. Surgical removal of tumor is preferred for MCS especially when tumor is accessible and can be completely excised. Similarly, radiation can be utilized for localized control, especially when surgically not accessible or in advanced disease for palliative approach to alleviate symptoms. Chemotherapy can be used to treat MCS either alone or in combination with other therapies. In case of aggressive systemic mastocytosis or mast cell leukemia, target therapy or even hematopoietic stem cell transplantation can be considered. Transient use of high-dose corticosteroid can also be utilized to decrease systemic MC burden [13].
Reports suggest that single-agent regimens such as bleomycin, vinblastine, or clofarabine, as well as combination approaches including anthracycline-based, cytarabine-based, or ifosfamide with cyclophosphamide and etoposide, and even AML-type protocols with or without fludarabine, have shown only modest effectiveness in treating MCS [13]. Furthermore, targeted therapies such as imatinib and dasatinib - tyrosine kinase inhibitors against KIT mutations - have been reported to be useful in prior literature, and midostaurin (PKC412) in combination with other therapies can be considered [35-37].
Conclusions
This case underscores the formidable diagnostic challenge posed by HLH, particularly when secondary to rare and aggressive malignancies such as MCS. The nonspecific presentation of HLH (fever, cytopenias, hepatosplenomegaly, and elevated inflammatory markers) often overlaps with other conditions, leading to delays in diagnosis and treatment. In patients presenting with FUO accompanied by cytopenias and organomegaly, clinicians should maintain a high index of suspicion for HLH, even in the absence of an immediately identifiable trigger.
This case also highlights the importance of early and comprehensive diagnostic evaluation, including prompt bone marrow biopsy and advanced molecular testing such as KIT mutation analysis, which may provide critical clues to an underlying neoplasm. Identifying malignancy-associated HLH early is essential, as outcomes are particularly poor without targeted therapy for the underlying disease process. Heightened clinical awareness, a multidisciplinary approach, and timely initiation of both HLH-directed therapy and malignancy-specific treatment may offer the best chance at improving outcomes in these fulminant and often fatal cases.
Acknowledgments
None to declare.
Financial Disclosure
Authors have no financial sources to disclose.
Conflict of Interest
Authors have no conflict of interest to disclose.
Informed Consent
Informed consent was obtained from family of the patient regarding this article publication.
Author Contributions
KJY: conceptualization, validation, supervision, writing - review and editing, methodology. AE: resources, writing - original draft. MR: methodology, writing - original draft. JU: methodology, writing - original draft. RM: writing - original draft. FP: writing - original draft. PPDT: writing - original draft. SS: validation, supervision, writing - review and editing. PA: validation, supervision, writing - review and editing, project administration.
Data Availability
All data related to this article is included within this article.
Abbreviations
HLH: hemophagocytic lymphohistiocytosis; MCS: mast cell sarcoma; IL-2: interleukin 2; ESR: erythrocyte sedimentation rate; CRP: C-reactive protein; NGS: next-generation sequencing; FUO: fever of unknown origin
| References | ▴Top |
- Hayden A, Park S, Giustini D, Lee AY, Chen LY. Hemophagocytic
syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: A systematic
scoping review. Blood Rev. 2016;30(6):411-420.
doi pubmed - Zhang R, Cui T, He L, Liu M, Hua Z, Wang Z, Wang Y. A study on early
death prognosis model in adult patients with secondary hemophagocytic lymphohistiocytosis.
J Healthc Eng. 2022;2022:6704859.
doi pubmed - Brito-Zeron P, Kostov B, Moral-Moral P, Martinez-Zapico A,
Diaz-Pedroche C, Fraile G, Perez-Guerrero P, et al. Prognostic factors of death in 151 adults
with hemophagocytic syndrome: etiopathogenically driven analysis. Mayo Clin Proc Innov Qual
Outcomes. 2018;2(3):267-276.
doi pubmed - Jumic S, Nand S. Hemophagocytic lymphohistiocytosis in adults:
associated diagnoses and outcomes, a ten-year experience at a single institution.
J Hematol. 2019;8(4):149-154.
doi pubmed - Parikh SA, Kapoor P, Letendre L, Kumar S, Wolanskyj AP. Prognostic
factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc.
2014;89(4):484-492.
doi pubmed - Otrock ZK, Eby CS. Clinical characteristics, prognostic factors, and
outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol.
2015;90(3):220-224.
doi pubmed - Kapoor S, Morgan CK, Siddique MA, Guntupalli KK. Intensive care unit
complications and outcomes of adult patients with hemophagocytic lymphohistiocytosis: A
retrospective study of 16 cases. World J Crit Care Med. 2018;7(6):73-83.
doi pubmed - Lehmberg K, Nichols KE, Henter JI, Girschikofsky M, Greenwood T,
Jordan M, Kumar A, et al. Consensus recommendations for the diagnosis and management of
hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica.
2015;100(8):997-1004.
doi pubmed - Daver N, McClain K, Allen CE, Parikh SA, Otrock Z, Rojas-Hernandez C,
Blechacz B, et al. A consensus review on malignancy-associated hemophagocytic
lymphohistiocytosis in adults. Cancer. 2017;123(17):3229-3240.
doi pubmed - Wang D, Tong X, Liu S, Zhang W, Wang L, Zhang S, Zhang T, et al.
Clinical characteristics and risk factors for 90-day overall survival among 204 adult patients
with secondary hemophagocytic lymphohistiocytosis: Experience from a single-center retrospective
study. Front Med (Lausanne). 2022;9:774959.
doi pubmed - Delavigne K, Berard E, Bertoli S, Corre J, Duchayne E, Demur C,
Mansat-De Mas V, et al. Hemophagocytic syndrome in patients with acute myeloid leukemia
undergoing intensive chemotherapy. Haematologica. 2014;99(3):474-480.
doi pubmed - Campo E, Harris NL, Pileri SA, Jaffe ES, Stein H, Thiele J. WHO classification of tumours of haematopoietic and lymphoid tissues [Internet]. IARC Who Classification of Tum; 2017. p. 586. Available from: https://books.google.com/books/about/WHO_Classification_of_Tumours_of_Haemato.html?hl=&id=Qdt9swEACAAJ.
- Monnier J, Georgin-Lavialle S, Canioni D, Lhermitte L, Soussan M,
Arock M, Bruneau J, et al. Mast cell sarcoma: new cases and literature review. Oncotarget.
2016;7(40):66299-66309.
doi pubmed - Weiler CR, Butterfield J. Mast cell sarcoma: clinical management.
Immunol Allergy Clin North Am. 2014;34(2):423-432.
doi pubmed - Petersdorf RG, Beeson PB. Fever of unexplained origin: report on 100
cases. Medicine (Baltimore). 1961;40:1-30.
doi pubmed - Durack DT, Street AC. Fever of unknown origin—reexamined and
redefined. Curr Clin Top Infect Dis. 1991;11:35-51.
pubmed - Arnow PM, Flaherty JP. Fever of unknown origin. Lancet.
1997;350(9077):575-580.
doi pubmed - Mulders-Manders C, Simon A, Bleeker-Rovers C. Fever of unknown
origin. Clin Med (Lond). 2015;15(3):280-284.
doi pubmed - Rosado FG, Kim AS. Hemophagocytic lymphohistiocytosis: an update on
diagnosis and pathogenesis. Am J Clin Pathol. 2013;139(6):713-727.
doi pubmed - Lee JC, Logan AC. Diagnosis and management of adult
malignancy-associated hemophagocytic lymphohistiocytosis. Cancers (Basel). 2023;15(6).
doi pubmed - Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S,
Ladisch S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic
lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131.
doi pubmed - Tamamyan GN, Kantarjian HM, Ning J, Jain P, Sasaki K, McClain KL,
Allen CE, et al. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: relation to
hemophagocytosis, characteristics, and outcomes. Cancer. 2016;122(18):2857-2866.
doi pubmed - Zoref-Lorenz A, Murakami J, Hofstetter L, Iyer S, Alotaibi AS,
Mohamed SF, Miller PG, et al. An improved index for diagnosis and mortality prediction in
malignancy-associated hemophagocytic lymphohistiocytosis. Blood. 2022;139(7):1098-1110.
doi pubmed - Georgin-Lavialle S, Aguilar C, Guieze R, Lhermitte L, Bruneau J,
Fraitag S, Canioni D, et al. Mast cell sarcoma: a rare and aggressive entity—report of
two cases and review of the literature. J Clin Oncol. 2013;31(6):e90-97.
doi pubmed - Mannelli F, Gesullo F, Mannarelli C, Vanderwert F, Lazzi S, Mungai F,
Berti V, et al. Diagnostic and therapeutic challenges in mast cell sarcoma.
Am J Hematol. 2023;98(3):529-532.
doi pubmed - Tefferi A, Abdelmagid M, Al-Kali A, Patnaik M, Hogan WJ, Begna K,
Gangat N, et al. Granularity in disease classification impacts survival prediction in advanced
systemic mastocytosis: A single institution study of 329 informative cases.
Am J Hematol. 2024;99(1):21-27.
doi pubmed - Colmenero I, Cozzolino I, Sotlar K, Alvarez-Twose I. Mast cell sarcoma. WHO classification of tumours online. 2025. Accessed January 27, 2025. https://tumourclassification.iarc.who.int/chaptercontent/63/21.
- Krystel-Whittemore M, Dileepan KN, Wood JG. Mast cell: a
multi-functional master cell. Front Immunol. 2015;6:620.
doi pubmed - Horny HP, Sotlar K, Valent P. Mastocytosis: immunophenotypical
features of the transformed mast cells are unique among hematopoietic cells. Immunol Allergy
Clin North Am. 2014;34(2):315-321.
doi pubmed - Singh A, Alkhateeb H, Pardanani A, He R, Orazi A, Tefferi A, Reichard
KK. Mast cell sarcoma: 2 Mayo Clinic cases. Am J Hematol.
2022;97(10):1381-1383.
doi pubmed - Geramizadeh B, Nabavizadeh S, Rezvani A, Shamsolvaezin N,
Zahedinassab A, Khodadadi N, Iranpour P. Mast cell sarcoma of small intestine, early diagnosis,
and good prognosis: an extremely rare case report and review of the literature. Gastrointest
Tumors. 2023;10(1):1-5.
doi pubmed - Collins N, Willard N, Pan Z. Mast cell sarcoma with KIT p.D816V
mutation and concurrent systemic mastocytosis. J Hematop. 2024;17(4):281-287.
doi pubmed - Arico M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G,
Martinetti M, et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the
International Registry. FHL Study Group of the Histiocyte Society. Leukemia.
1996;10(2):197-203.
pubmed - Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I
treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041-4052.
doi pubmed - Auquit-Auckbur I, Lazar C, Deneuve S, Guillemet C, Cordel N,
Blanchard F, Joly P, et al. Malignant transformation of mastocytoma developed on skin
mastocytosis into cutaneous mast cell sarcoma. Am J Surg Pathol.
2012;36(5):779-782.
doi pubmed - Ryan RJ, Akin C, Castells M, Wills M, Selig MK, Nielsen GP, Ferry JA,
et al. Mast cell sarcoma: a rare and potentially under-recognized diagnostic entity with
specific therapeutic implications. Mod Pathol. 2013;26(4):533-543.
doi pubmed - Bautista-Quach MA, Ake CD, Chen M, Wang J. Gastrointestinal
lymphomas: Morphology, immunophenotype and molecular features. J Gastrointest Oncol.
2012;3(3):209-225.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Journal
of Hematology is published by Elmer Press Inc.