| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://jh.elmerpub.com |
Original Article
Volume 000, Number 000, July 2025, pages 000-000

Novel Red Blood Cell Exchange Parameters for Treatment of Transfusion-Dependent Thalassemia Based on Experience of Five Patients
Kristina Sevcika, c, Claire Jacksonb, c, Shelly M. Williamsa, Scott A. Koepsella, Aleh Bobra, d
aDepartment of Pathology, Microbiology and Immunology, University of Nebraska Medical Center, Omaha, NE, USA
bDepartment of Pediatrics, University of Nebraska Medical Center, Omaha, NE, USA
cThese authors contributed equally to this work.
dCorresponding Author: Aleh Bobr, Department of Pathology, Microbiology and Immunology, University of Nebraska Medical Center, Omaha, NE, USA
Manuscript submitted May 16, 2025, accepted June 26, 2025, published online July 8, 2025
Short title: RBCX for Treatment of TDT
doi: https://doi.org/10.14740/jh2086
| Abstract | ▴Top |
Background: Thalassemias are inherited red blood cell disorders characterized by defective globin production, resulting in microcytic hypochromic anemia. Severe variants lead to transfusion dependence and consequent iron overload, often despite chelation therapy. The role of automated red blood cell exchange (RBCX) for transfusion-dependent thalassemia (TDT) is unclear and previously there was no specific apheresis parameters specific for thalassemia defined. We present our experience with patients with TDT treated with RBCX using higher hematocrit parameters specifically tailored for this condition.
Methods: Five patients with TDT underwent chronic RBCX in place of simple transfusion with the primary goal of stabilizing iron overload. Novel parameters were established to satisfy the Thalassemia International Federation goal pre-transfusion hemoglobin of 9.5 g/dL, while targeting a post-transfusion hematocrit of 37-38%.
Results: RBCX was well tolerated with infrequent occurrences of vasovagal reactions, asymptomatic hypotension, citrate side effects, and vascular access issues. The transfusion interval increased from an average of 3 weeks with simple transfusions to 5 weeks with RBCX. Despite an increase in average blood utilization, serum ferritin levels remained stable.
Conclusion: RBCX with higher hematocrit parameters can be performed safely and efficiently in TDT patients. To our knowledge, this is the first report of TDT-specific RBCX parameters. Though blood utilization is higher with RBCX, it offers longer intervals between transfusions and has no increase in iron overload, improving quality of life for patients.
Keywords: Beta-thalassemia; RBC exchange; Iron overload
| Introduction | ▴Top |
Patients with transfusion-dependent thalassemia (TDT) are commonly treated with frequent simple transfusions, typically preformed every 2 - 4 weeks to suppress ineffective erythropoiesis and alleviate anemia [1, 2]. Iron overload (as a complication of transfusions) can lead to serious comorbidities related to iron deposition in the liver, pancreas, and heart [2]. Iron chelators currently indicated for management of iron overload in TDT include deferoxamine, deferiprone, and deferasirox [3-5]. Red blood cell exchange (RBCX) has been used in the treatment of sickle cell disease (SCD), offering the dual benefits of removing defective or senescent red blood cells (RBCs) and minimizing iron overload. For TDT, there is a paucity of literature evaluating RBCX as a chronic treatment modality, especially using hematocrit targets that reflect international guidelines [6].
| Materials and Methods | ▴Top |
Between 2019 and 2021, five patients with TDT were transitioned from chronic simple RBC transfusions to RBCX after developing iron overload as evidenced by serum ferritin elevation with use of deferasirox, with a ferritin reference range of 23 - 340 ng/mL for males and 11 - 310 ng/mL for females. At the time of RBCX initiation, patient A was an 11-year-old male (beta-thalassemia major) with a peak ferritin of 7,077 ng/mL and T2-weighted magnetic resonance imaging (MRI) suggestive of iron deposition. Patient B was a 19-year-old male (beta-thalassemia major) with a peak ferritin of 1,310 ng/mL and deferasirox use complicated by mild hearing loss. Patient C was a 14-year-old male (beta-thalassemia/HbE) with a peak ferritin of 1,400 ng/mL and deferasirox management complicated by nephrotoxicity. Patient D was a 15-year-old male (beta-thalassemia major) with a peak ferritin of 3,060 ng/mL. Patient E was a 14-year-old male (beta-thalassemia/HbE) with a peak ferritin of 3,404 ng/mL who also experienced chelation-related nephrotoxicity.
The study has been approved by IRB. Since the study is a retrospective review of data, the informed consent is not required.
The patients underwent RBCX with new parameters for TDT that we developed to satisfy international guidelines [7], while continuing to optimize chelation therapy. Procedures were performed on the Terumo BCT Spectra Optia Apheresis System, with donor blood matched for Rh (D, C/c, E/e) and Kell blood antigens to minimize risk of RBC alloimmunization. Routine complete blood count (CBC) and ferritin levels were obtained before each procedure, as was a post-procedure CBC. Additional data collection included transfusion frequency, chelation therapy, volume of blood administered, and any adverse events. The procedure parameters and intervals were adjusted based on hematocrit levels and tolerance of procedure.
Transfusion intervals were individualized based on the patient’s pre-transfusion hematocrit levels to target a pre-transfusion hematocrit of 29% or higher, based on the Thalassemia International Federation guidelines of transfusing to maintain hemoglobin above a level of 9.5 to suppress ineffective erythropoiesis and its sequelae [7]. The post-transfusion hematocrit targets were set to 37-38%, allowing maintained or extended transfusion intervals before reaching the higher pre-transfusion target. This is in contrast to the recommended maximum hematocrit in SCD, in which the increased viscosity of higher hematocrits may precipitate sickling. For several patients, a post-RBCX hematocrit of 40% was attempted with an increased rate of vasovagal reactions observed. The fraction of cells remaining (FCR) was set to 30%. To preserve blood products with the increased hematocrit goals, isovolemic hemodilution was not performed. Performing isovolemic hemodilution with FCR30% was not possible by machine settings. To achieve high post-transfusion hematocrit target with isovolemic dilution performed, the machine required lower FCR and higher blood volume to use. The hematocrit of transfused units was assumed to be at 60%. As summarized in Table 1, these RBCX settings differ from those commonly used for treatment of SCD.
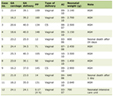 Click to view | Table 1. RBCX Parameters Utilized for the Described TDT Patients Versus Previously Described Exchange Parameters Utilized for Patients With SCD |
Data were retrospectively analyzed after several years of RBCX. Ferritin levels from 1 year prior to RBCX and the subsequent 2 years of receiving RBCX were analyzed using a Wilcoxon signed-rank test to assess the primary endpoint of controlling iron overload, with analysis conducted in SAS version 9.4. The volume of transfused blood was compared between the 3 months prior to and after initiation of RBCX. All procedures performed up to the time of analysis were evaluated for adverse reactions.
| Results | ▴Top |
The patients underwent RBCX over a range of 2 - 4 years, encompassing a total of 188 procedures at the time of analysis. Of these, 17 vasovagal reactions were observed, which predominately occurred with post-transfusion hematocrits of 40% or higher. Additionally, eight asymptomatic hypotensive reactions were noted during routine vital sign monitoring, and four citrate toxicity reactions occurred that were abrogated with co-infusion of intravenous calcium gluconate. No other transfusion or apheresis reactions were observed.
The procedures (despite higher blood utilization) were effective in stabilizing iron overload in conjunction with iron chelation therapy as described in Table 2. The median pre-RBCX ferritin level for the study group was 2,489.3 ng/mL, and the median post-RBCX ferritin level was 1,329.3 ng/mL. After subtracting the post-RBCX ferritin level from the pre-RBCX ferritin level for each patient, the median change for the group was 457 ng/mL (P = 0.06; P < 0.05 is considered statistically significant). The trends of ferritin for the individual patients are shown in Figure 1. The ferritin comparison is done.
Click to view | Table 2. Mean Ferritin Levels, Blood Utilization, and Transfusion Intervals for Each Patient |
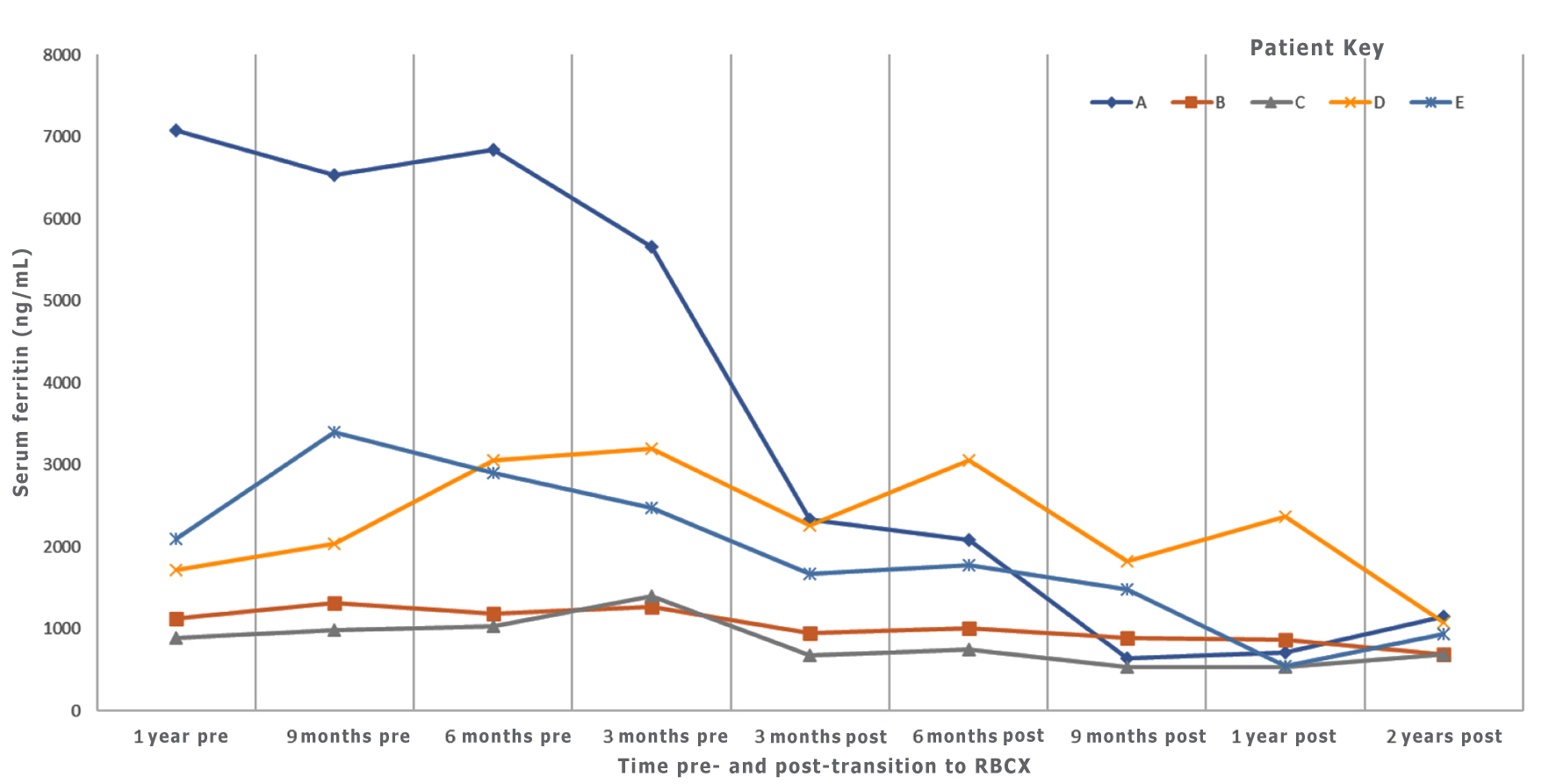 Click for large image | Figure 1. Serum ferritin level trends pre- and post-transition to RBCX. Serum ferritin levels were trended for patients at 3-month intervals which showed ferritin stability or improvement as patients continued RBCX. RBCX: red blood cell exchange. |
The patients were largely able to maintain similar deferasirox doses while on RBCX. At the time of initiating RBCX, patient A was receiving 720 mg/day, which was increased to 900 mg/day 2 years later. Patient B remained on 1,080 mg/day. Patient C was on 360 mg/day and discontinued chelation therapy by the 2-year time point due to ongoing nephrotoxicity. Patient D increased from 900 to 1,080 mg/day. Patient E increased from 360 to 1,080 mg/day.
RBCX required increased blood product utilization compared to simple transfusion. The average blood use per patient was 4,085 mL more over a 3-month period with RBCX. However, the transfusion interval also increased for each patient, as described in Table 2. The average time between simple transfusions was 3.2 weeks, compared to 5 weeks after transitioning to RBCX. Despite increased exposure to RBC units, none of these patients have developed RBC antibodies while on RBCX to date.
| Discussion and Conclusion | ▴Top |
Current guidelines have established the utility of RBCX primarily for various states of SCD, including acute stroke and stroke prophylaxis as the only first-line indications, and as second-line therapy for acute chest syndrome and liver disease in erythropoietic protoporphyria. While RBCX has been used for many other conditions (including TDT), its role remains unclear and decision-making is individualized [8]. While no established guidelines exist for RBCX in thalassemia, several studies have reported its effective use in thalassemic patients for reducing hemoglobin F levels, thereby promoting wound healing [3, 9] or alleviating respiratory distress from cor pulmonale [10]. Here in this paper, we do not address efficiency and clinical utility of RBCX in thalassemia; the case series is not an appropriate study for that. Rather, we suggest that if individualized decision is made to pursue RBCX in these patients, one can safely utilize the RBCX parameters discussed here in the article to comply with internation guidelines [7].
This case series found that when higher hematocrit targets are used for RBCX, complication rates are low, RBC alloantibody formation is not increased, ferritin levels are stabilized or decreased, and time in between transfusions is increased. These findings contrast with a recent study suggesting no utility for RBCX in place of simple transfusions for the same type of TDT [11]. Our approach differs significantly in that the previous study used RBCX hematocrit levels appropriate for SCD. We believe that the different outcomes are due to the use of higher hematocrit RBCX targets appropriate for the use of thalassemia, providing the dual benefit of suppressing ineffective erythropoiesis and partially removing old, previously transfused erythrocytes to control iron overload.
The observed ferritin levels in all patients show an overall downward trend, though not statistically significant, despite increased blood utilization. It is also notable that patient C, who experienced chelation-related nephrotoxicity, was able to discontinue chelation entirely and manage iron overload with RBCX alone, achieving reduced ferritin levels compared to pre- RBCX values. Our experience can potentially support the use of RBCX to stabilize iron overload and offers a potentially novel option for patients who are refractory to or intolerant of chelation therapy. We hypothesize that the mechanism of iron overload stabilization via RBCX is by removing senescent autologous or previously transfused cells from circulation before they can be taken up by reticuloendothelial macrophages and add to the iron deposits.
We demonstrate that RBCX can be considered safe and has a possibility to be an effective management for TDT with iron overload, offering the additional benefit of reduced procedure burden. A drawback to this approach is the increased blood utilization, which if broadly applied to patients with TDT, could present supply challenges. This needs to be taken in the account, especially if the treating institution experiences frequent “blood shortages”. We also understand that the results of this case series are based on a small number of patients and need to be interpreted with caution hoping that additional data in the future will be able to clarify the clinical efficacy of this approach.
Acknowledgments
We acknowledge the nursing staff of Apheresis and Pediatric Hematology at Nebraska Medicine and Children’s Nebraska for tremendous help in organizing care for the patients. Additional thanks for the Nebraska Medicine Hospital Blood Bank and Nebraska Community Blood Bank for their help in providing blood supply for the patients described.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Not applicable.
Author Contributions
Kristina Sevcik and Claire Jackson: analysis of data and writing of manuscript. Shelly M. Williams and Scott A. Koepsell: analysis of data and editorial review of manuscript. Aleh Bobr: analysis of data, writing the manuscript, and developing idea of the paper.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Coates TD. Iron overload in transfusion-dependent patients. Hematology Am Soc Hematol Educ Program. 2019;2019(1):337-344.
doi pubmed - Farmakis D, Porter J, Taher A, Domenica Cappellini M, Angastiniotis M, Eleftheriou A. 2021 Thalassaemia International Federation Guidelines for the management of transfusion-dependent thalassemia. Hemasphere. 2022;6(8):e732.
doi pubmed - Pignatti M, Govoni M, Graldi G, Pacchioni L, De Santis G, Borgna C. Thalassaemia intermedia: the role of erythroexchange in the treatment of an indolent wound. Blood Transfus. 2014;12(1):124-126.
doi pubmed - Agarwal MB, Gupte SS, Viswanathan C, Vasandani D, Ramanathan J, Desai N, Puniyani RR, et al. Long-term assessment of efficacy and safety of L1, an oral iron chelator, in transfusion dependent thalassaemia: Indian trial. Br J Haematol. 1992;82(2):460-466.
doi pubmed - Cappellini MD, Cohen A, Piga A, Bejaoui M, Perrotta S, Agaoglu L, Aydinok Y, et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood. 2006;107(9):3455-3462.
doi pubmed - Berdoukas VA, Kwan YL, Sansotta ML. A study on the value of red cell exchange transfusion in transfusion dependent anaemias. Clin Lab Haematol. 1986;8(3):209-220.
doi pubmed - Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V. Guidelines for the management of transfusion dependent thalassaemia (TDT). 3rd ed. Nicosia (CY); 2014.
pubmed - Connelly-Smith L, Alquist CR, Aqui NA, Hofmann JC, Klingel R, Onwuemene OA, Patriquin CJ, et al. Guidelines on the use of therapeutic apheresis in clinical practice - evidence-based approach from the Writing Committee of the American Society for Apheresis: the ninth special issue. J Clin Apher. 2023;38(2):77-278.
doi pubmed - Aessopos A, Kati M, Tsironi M, Polonifi E, Farmakis D. Exchange blood transfusions for the treatment of leg ulcerations in thalassemia intermedia. Haematologica. 2006;91:(5 Suppl):ECR11.
pubmed - Kopparthy P, Kelkar AH, Aggarwal K, De Filippis S, Fletcher B. Red blood cell exchange in a patient with extramedullary hematopoiesis and cor pulmonale secondary to beta thalassemia. Cureus. 2021;13(3):e13638.
doi pubmed - Wall E, Bolster L. Complications of red cell exchange for anemia management in patients with transfusion-dependent thalassemia. Transfusion. 2023;63(7):1277-1283.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.