| Journal of Hematology, ISSN 1927-1212 print, 1927-1220 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Hematol and Elmer Press Inc |
| Journal website https://jh.elmerpub.com |
Case Report
Volume 000, Number 000, April 2025, pages 000-000

Amyloid Light Chain Amyloidosis Masquerading as Scleroderma: A Diagnostic Challenge
Kriti Dhamijaa, c ![]() , Rahim A. Jiwanib, Arjun Lakshamanb, Santhosh Sadashivb, Prerna Mewawallab
, Rahim A. Jiwanib, Arjun Lakshamanb, Santhosh Sadashivb, Prerna Mewawallab
aMedicine Institute, Allegheny Health Network, Pittsburgh, PA 15212, USA
bDivision of Hematology and Cellular Therapy, Allegheny Health Network Cancer Institute, Allegheny Health Network, Pittsburgh, PA 15212, USA
cCorresponding Author: Kriti Dhamija, Medicine Institute, Allegheny Health Network, Pittsburgh, PA 15212, USA
Manuscript submitted March 12, 2025, accepted April 12, 2025, published online April 22, 2025
Short title: AL Amyloidosis Masquerading as Scleroderma
doi: https://doi.org/10.14740/jh2055
| Abstract | ▴Top |
Systemic amyloidosis has diverse, often nonspecific, clinical manifestations that overlap or mimic other medical disorders, making amyloidosis a diagnostic challenge. We present a case of a middle-aged female who presented with skin thickening, fatigue, arthritis, and macroglossia, which were initially thought to be due to systemic sclerosis. With no response to immunosuppressive therapies, she was tested for plasma cell dyscrasias. Additional work-up and cardiac biopsy were positive for amyloid light chain (AL) amyloidosis. The diagnosis was delayed by 2 years because the protein electrophoresis ordered at the initial encounter was not accompanied by serum-free light chain testing. This case emphasizes the importance of considering amyloidosis in patients with unexplained systemic symptoms and highlights the role of a comprehensive diagnostic evaluation.
Keywords: Amyloidosis; Smoldering multiple myeloma; Congo red staining; Free light chain
| Introduction | ▴Top |
Amyloidosis is a group of rare diseases marked by the abnormal folding of proteins into insoluble beta-pleated sheets that are deposited extracellularly into various organs impairing normal organ function. Although amyloid light chain (AL) amyloidosis remains the most common form of amyloidosis, the incidence of wild-type transthyretin amyloidosis (ATTR) has also been increasing [1, 2]. In a United Kingdom (UK)-based study in 2013, 65% of all amyloidosis cases diagnosed were labeled as AL amyloidosis [3]. However, in a more recent Germany-based national registry data from 2018 - 2020, 45.5% of all amyloidosis cases were AL amyloidosis and 39.6% were ATTR cases [4]. Despite being the most common form, it remains an extremely rare disease, with incidence ranging from 9.7 to 14.0 cases per million person years, as reported in a US data-based study from 2007 to 2015 [5, 6]. AL amyloidosis arises from a plasma cell disorder where monoclonal light chains aggregate into amyloid fibrils [5]. These fibrils can infiltrate virtually any organ outside the central nervous system, manifesting a wide range of clinical symptoms [5].
The diagnosis of AL amyloidosis is often delayed due to its non-specific symptoms and clinical overlap with other disorders, such as systemic sclerosis (scleroderma) [7]. The hallmark of amyloidosis is the presence of amyloid fibrils, which can be identified as pink extracellular deposits in the organ biopsy with hematoxylin and eosin (H&E) stain and by apple-green birefringence under polarized light on Congo red staining. However, routine laboratory tests may be inconclusive, especially in cases where monoclonal proteins are not detected in a patient’s serum or urine using protein electrophoresis or immunofixation. The serum free light chain (FLC) assay plays a critical role in these cases, as around 15% of patients with primary AL amyloidosis do not exhibit monoclonal protein in serum or urine electrophoresis [8].
We report a 54-year-old woman with AL amyloidosis who was initially misdiagnosed as having systemic sclerosis, highlighting the diagnostic challenges posed by this condition.
| Case Report | ▴Top |
Investigations
A 54-year-old woman presented with skin thickening, pruritus, fatigue, arthralgias, macroglossia, and Raynaud’s phenomenon. She had a prior history of ulcerative colitis and rectal adenocarcinoma that was treated with surgical resection and chemotherapy. The patient’s arthritis was most pronounced in the neck, right shoulder, wrists, and fingers, with evidence of Bouchard and Heberden nodes. The combination of Raynaud’s phenomenon, skin changes, and arthritis led to a provisional diagnosis of systemic sclerosis.
Initial serologic testing was negative, including antinuclear antibody, scleroderma-specific autoantibodies (Scl-70), and a panel for other rheumatologic markers (ribonucleoprotein, RNA polymerase III, rheumatoid factor, and C-reactive protein). A specialized antibody panel (Oklahoma scleroderma panel from the Oklahoma Medical Research Foundation in Oklahoma City, OK) was used to screen for antibodies involved in different autoimmune diseases like lupus, mixed connective tissue disease, Sjogren’s syndrome, and scleroderma. Despite the negative results, there was still clinical suspicion of systemic sclerosis, so the patient was treated with a variety of immunosuppressive therapies, including prednisone, methotrexate, mycophenolate mofetil, tocilizumab, and rituximab. The patient’s symptoms did not improve. As part of the testing on the first encounter, a serum protein electrophoresis (SPEP) was obtained, which showed hypogammaglobulinemia with no identifiable monoclonal protein, but no immunofixation or FLC assays were performed.
Diagnosis
Two years after receiving the negative test results and failing to respond to immunosuppression, the patient developed a rash over the chin described as sclerosis with telangiectasias. Her new dermatologic findings raised suspicion of advanced systemic sclerosis, and her immunosuppressive treatment was continued, with consideration of more aggressive additional systemic therapy for scleroderma and topical tacrolimus ointment. However, given the patient’s persistent symptoms and lack of response to immunosuppressive therapy, repeated investigations were performed. SPEP and serum immunofixation did not reveal a monoclonal protein. FLCs were also checked this time; the results showed lambda light chains elevated to 36.58 mg/dL and kappa light chains at 0.5 mg/dL, with an involved to uninvolved light chain ratio of 73 (normal range for kappa to lambda light chain ratio is 0.26 - 1.65). A subsequent bone marrow biopsy revealed a plasma cell population comprising 30% of the bone marrow cellularity, and in situ hybridization confirmed the plasma cells were lambda light chain restricted. The Congo red staining of the bone marrow was negative for amyloidosis. A whole-body magnetic resonance imaging (MRI) of the patient’s bone marrow supply showed a partially imaged right distal femoral T2 hyperintense lesion centered at the physeal scar, which was further characterized as not being a myeloma-defining bone lesion with contrast-enhanced MRI of the knee. The patient had no anemia, hypercalcemia, or renal injury.
She did not have symptoms of dyspnea on exertion, orthopnea, or paroxysmal nocturnal dyspnea, palpitations, chest pain, or leg swelling. However, due to suspicion of underlying AL amyloidosis, her history of macroglossia, and the FLC findings, an echocardiogram with strain imaging was performed. The echocardiogram revealed moderate concentric hypertrophy of the left ventricle, which is consistent with infiltrative cardiomyopathy and was confirmed with a cardiac MRI. Pro-B-type natriuretic peptide (Pro-BNP) was measured as 291 pg/mL, and high-sensitivity troponin was 14 ng/L at the time. Twenty-four-hour urine protein was slightly elevated at 331.3 mg/24 h. An endomyocardial biopsy was performed (Fig. 1) and showed positive staining on Congo red (Fig. 2), and liquid chromatography/mass spectrometry confirmed AL amyloidosis. She was thus diagnosed with Mayo stage II AL amyloidosis [9]. The patient did not have any other symptoms suggestive of amyloidosis, such as peripheral neuropathy, autonomic dysregulation, hepatosplenomegaly, or bleeding diathesis. She was deemed to have a poor prognosis due to bone marrow involvement with ≥ 10% plasma cells [10].
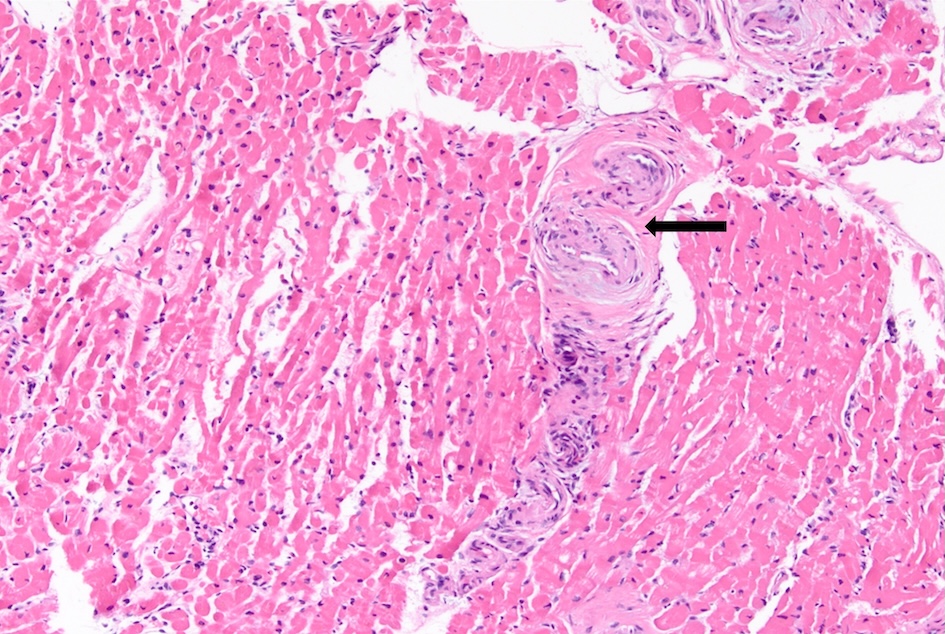 Click for large image | Figure 1. Hematoxylin and eosin stain on the patient’s bone marrow biopsy showing amorphous eosinophilic deposits (black arrow). |
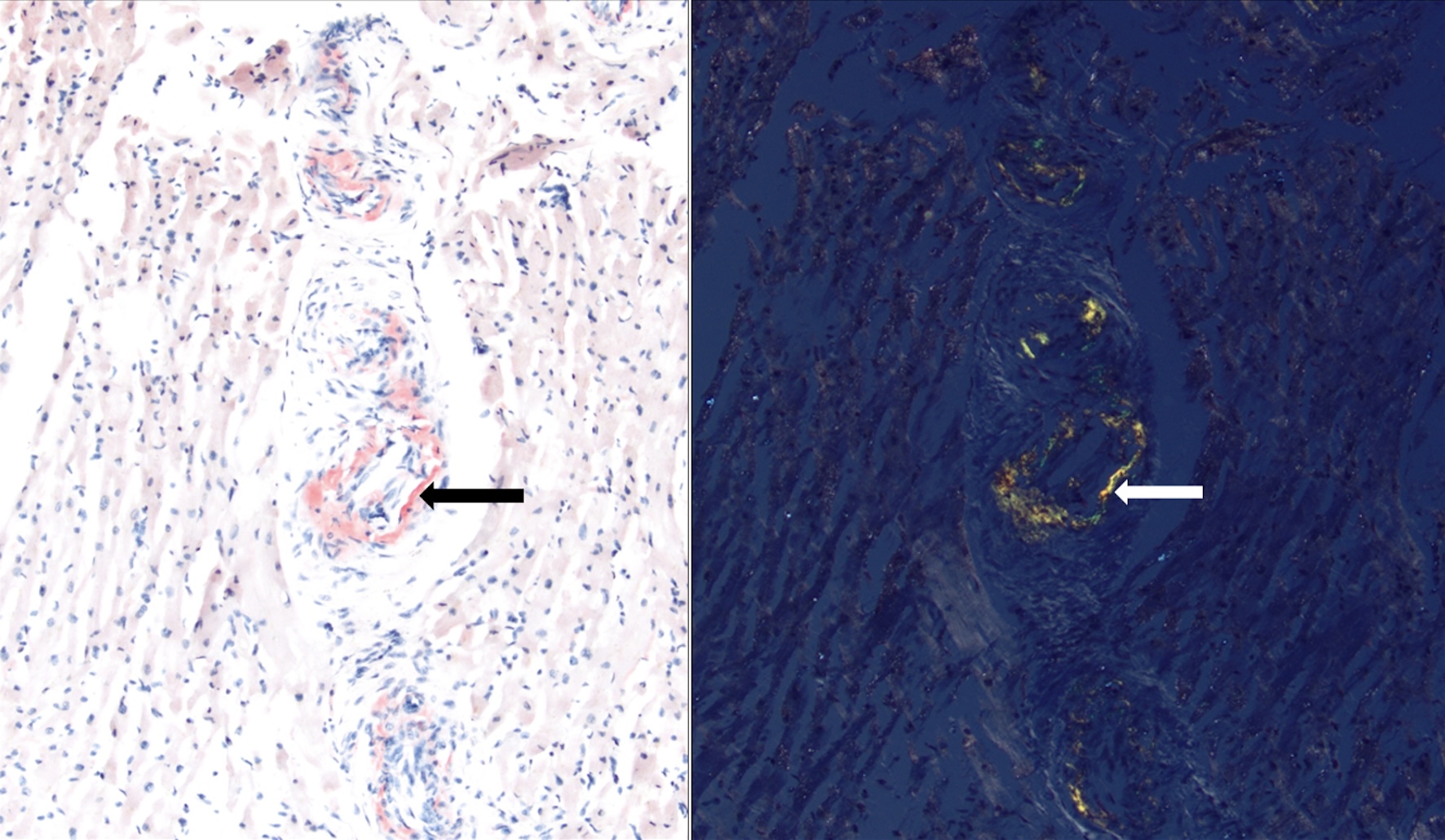 Click for large image | Figure 2. Native heart endomyocardial biopsy showing mild intramyocardial arterial mural involvement, staining positive for Congo red stain under normal light (left; red staining, black arrow) and polarized light (right; apple-green birefringence, white arrow). |
Treatment and outcomes
She was treated with daratumumab, cyclophosphamide, bortezomib, and dexamethasone as recommended by the ANDROMEDA clinical trial [11] with complete hematological and renal response after six cycles of therapy. She did not have any cardiac response. She underwent consolidation with a high dose of melphalan 200 mg/m2, followed by autologous stem cell rescue. At approximately 1 year and 8 months after autologous stem cell transplantation, she remains on single-agent daratumumab maintenance therapy in complete hematological remission. Her prior symptoms completely resolved, including skin changes. Her arthralgias resolved, and her macroglossia improved by 50%.
| Discussion | ▴Top |
We present a middle-aged female who presented with skin thickening, fatigue, arthritis, and macroglossia. Being initially diagnosed with systemic sclerosis, her symptoms did not show any improvement with usual scleroderma-directed therapies. Two years later, she tested positive for smoldering multiple myeloma. During the course of underlying multiple myeloma, 12% to 15% of patients manifest clinically evident amyloidosis [12]. In a 200-patient-based study done by Vela-Ojeda et al [13], 34% of patients with multiple myeloma were diagnosed with amyloidosis eventually; 8% were subclinical cases, 18% of cases progressed to develop clinical amyloidosis during the course of the disease, and another 8% had signs and symptoms of amyloidosis at the time of presentation [13].
Our patient had non-specific mucocutaneous findings on presentation, which made the diagnosis challenging and delayed. In amyloidosis, skin involvement is seen in 30% to 40% of cases, and the associated symptoms can overlap with autoimmune conditions like systemic sclerosis, leading to diagnostic confusion (Table 1 [14-21]). Cutaneous and mucosal involvement (Table 2) can lead to amyloidosis being mistaken for scleroderma [22], since scleroderma shares symptoms of skin thickening/tightness, Raynaud’s phenomenon, and even gastrointestinal involvement [23]. However, macroglossia, being a classical sign, increased the suspicion of amyloidosis in our patient.
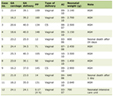 Click to view | Table 1. Symptoms Observed in Different Organs and Organ Systems for Amyloidosis Compared to Scleroderma |
Click to view | Table 2. System-Specific Symptoms of Amyloidosis Compared to Scleroderma |
AL amyloidosis has a wide range of clinical presentations dependent on the organ-specific deposition of amyloid fibrils. The nature of organ involvement depends on the specific gene mutations in the light chain region of the immunoglobulins. These gene mutations cause decreased stability and heightened protein dynamics, leading to the formation of soluble oligomers and amyloid fibrils. For example, the germline gene LV6-57 is more prone to involve the kidneys, whereas LV1-44 has a predilection for heart involvement [24].
Another important aspect of our case was that the delay in diagnosis could have been minimized by a comprehensive approach to diagnosis. In addition to overlapping symptoms making diagnosis difficult, standard laboratory tests may fail to detect monoclonal proteins in up to 50% of amyloidosis cases [9]. This underscores the importance of employing serum FLC assays, especially in cases with abnormal clinical features and unexplained laboratory results. When there is suspicion of amyloidosis, the initial work-up should include labs like serum FLC assay, SPEP, and serum immunofixation. Cardiac involvement should be investigated with high-sensitivity troponin, N-terminal pro-BNP, and echocardiography with strain imaging. A cardiac MRI can also be considered. In case of an M (monoclonal protein) spike or abnormal FLC ratio, bone marrow biopsy and/or abdominal fat pad aspiration should be done with Congo red staining. If negative, and amyloidosis suspicion persists, an organ biopsy should also be performed, as bone marrow and fat pad biopsy can be negative in 20% of cases [25].
Once the diagnosis of systemic amyloidosis has been established, the next step is to type the amyloid by determining the amyloid protein involved. Mass spectrometry-based proteomic analysis of subcutaneous fat pad aspirates, which has a sensitivity of 88%, a specificity of 96%, and an accuracy of 91%, is used to analyze and classify the formalin-fixed paraffin-embedded tissues [26].
The prognosis of AL amyloidosis is significantly affected by cardiac involvement, and hence, the disease is staged based on the value of pro-BNP, troponin T, and the difference between involved and uninvolved FLC [27]. Approximately 50% of patients present with heart failure symptoms, such as dyspnea, orthopnea, and edema. In this case, the absence of overt cardiac symptoms initially delayed the suspicion of amyloidosis, further complicating the diagnostic process. However, the persistence of macroglossia, an unusual but characteristic feature of amyloidosis, prompted further investigation and led to the correct diagnosis.
Diagnosing AL amyloidosis requires a high degree of clinical suspicion, especially in patients with nonspecific, progressive symptoms. Once amyloidosis is suspected, tissue biopsy remains the gold standard for diagnosis, with Congo red staining being highly specific for amyloid deposits. In cases where monoclonal proteins are not detectable, mass spectrometry-based proteomics can be used to definitively type the amyloid and guide appropriate treatment.
Learning points
This case illustrates the diagnostic complexity of amyloidosis and emphasizes the importance of considering this rare condition in the differential diagnosis of patients presenting with systemic symptoms, particularly when autoimmune and rheumatologic etiologies have been inconclusive. Macroglossia is highly suggestive of AL amyloidosis. In patients with suspected systemic amyloidosis, a multidisciplinary approach involving hematology, cardiology, and neurology is required for timely diagnosis and management with a comprehensive diagnostic approach, including FLC assays, bone marrow biopsy, and cardiac imaging. Increased clinical awareness of amyloidosis in patients with overlapping features of systemic sclerosis may prevent delayed diagnoses, reduce morbidity, and improve patient outcomes.
Acknowledgments
The authors thank Sarah Carey, MS, Jade Chang, and Jacalyn Newman, PhD, of Allegheny Health Network’s Health System Publication Support Office (HSPSO) for their assistance in editing and formatting the manuscript. The HSPSO is funded by Highmark Health (Pittsburgh, PA, United States of America), and all work was done in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images.
Author Contributions
Kriti Dhamija participated in writing and editing the case report. Rahim Jiwani, Arjun Lakshaman, Santhosh Sadashiv, and Prerna Mewawalla participated in editing the report. All the authors read and approved the final version of the manuscript.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
Abbreviations
AL: amyloid light chain; ANCA: anti-neutrophil cytoplasmic antibody; FLC: free light chain; GERD: gastroesophageal reflux disease; MRI: magnetic resonance imaging; pro-BNP: pro-brain natriuretic peptide; SPEP: serum protein electrophoresis
| References | ▴Top |
- Picken MM. The pathology of amyloidosis in classification: a review. Acta Haematol. 2020;143(4):322-334.
doi pubmed - Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387(10038):2641-2654.
doi pubmed - Pinney JH, Smith CJ, Taube JB, Lachmann HJ, Venner CP, Gibbs SD, Dungu J, et al. Systemic amyloidosis in England: an epidemiological study. Br J Haematol. 2013;161(4):525-532.
doi pubmed - Hegenbart U, Siepen Fad, Carpinteiro A, Hansen T, Kimmich C, Oldenburg S, Hofmann E, et al. Long-term evaluation of amyloidosis diseases in germany: national clinical amyloidosis registry. Amyloid-Journal of Protein Folding Disorders. 2024;31:S147-S147.
doi - Dima D, Mazzoni S, Anwer F, Khouri J, Samaras C, Valent J, Williams L. Diagnostic and treatment strategies for AL amyloidosis in an era of therapeutic innovation. JCO Oncol Pract. 2023;19(5):265-275.
doi pubmed - Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv. 2018;2(10):1046-1053.
doi pubmed - Dziadzio M, Anastassiades CP, Hawkins PN, Potter M, Gabrielli A, Brough GM, Black CM, et al. From scleredema to AL amyloidosis: disease progression or coincidence? Review of the literature. Clin Rheumatol. 2006;25(1):3-15.
doi pubmed - Chau EM, Cheung SC, Chow SL, Fu KH. Nonsecretory immunoglobulin-derived amyloidosis of the heart: diagnosis by immunohistochemistry of the endomyocardium. Clin Cardiol. 1997;20(5):494-496.
doi pubmed - Kumar SK, Callander NS, Adekola K, Anderson LD, Jr., Baljevic M, Campagnaro E, Castillo JJ, et al. Systemic light chain amyloidosis, Version 2.2023, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2023;21(1):67-81.
doi pubmed - Gustine JN, Staron A, Mendelson L, Joshi T, Gopal DM, Siddiqi OK, Ruberg FL, et al. Predictors of treatment response and survival outcomes in patients with advanced cardiac AL amyloidosis. Blood Adv. 2023;7(20):6080-6091.
doi pubmed - Kastritis E, Palladini G, Minnema MC, Wechalekar AD, Jaccard A, Lee HC, Sanchorawala V, et al. Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N Engl J Med. 2021;385(1):46-58.
doi pubmed - Bahlis NJ, Lazarus HM. Multiple myeloma-associated AL amyloidosis: is a distinctive therapeutic approach warranted? Bone Marrow Transplant. 2006;38(1):7-15.
doi pubmed - Vela-Ojeda J, Garcia-Ruiz Esparza MA, Padilla-Gonzalez Y, Sanchez-Cortes E, Garcia-Chavez J, Montiel-Cervantes L, Reyes-Maldonado E, et al. Multiple myeloma-associated amyloidosis is an independent high-risk prognostic factor. Ann Hematol. 2009;88(1):59-66.
doi pubmed - Shanmugam VK, Steen VD. Renal disease in scleroderma: an update on evaluation, risk stratification, pathogenesis and management. Curr Opin Rheumatol. 2012;24(6):669-676.
doi pubmed - Taiwo AA, Alapati L, Movahed A. Cardiac amyloidosis: a case report and review of literature. World J Clin Cases. 2019;7(6):742-752.
doi pubmed - Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008;34(1):181-190.
doi pubmed - Wetwittayakhlang P, Sripongpun P, Jandee S. Primary gastrointestinal amyloidosis: an unusual cause of acute intestinal pseudo-obstruction. Case Rep Gastroenterol. 2019;13(3):462-467.
doi pubmed - Shreiner AB, Murray C, Denton C, Khanna D. Gastrointestinal manifestations of systemic sclerosis. J Scleroderma Relat Disord. 2016;1(3):247-256.
doi pubmed - Gandham AK, Gayathri AR, Sundararajan L. Pulmonary amyloidosis: a case series. Lung India. 2019;36(3):229-232.
doi pubmed - Fotiou D, Dimopoulos MA, Kastritis E. Systemic AL amyloidosis: current approaches to diagnosis and management. Hemasphere. 2020;4(4):e454.
doi pubmed - Frech TM, Mar D. Gastrointestinal and hepatic disease in systemic sclerosis. Rheum Dis Clin North Am. 2018;44(1):15-28.
doi pubmed - Sun L, Zhang L, Hu W, Li TF, Liu S. Case report: One case of primary AL amyloidosis repeatedly misdiagnosed as scleroderma. Medicine (Baltimore). 2017;96(50):e8771.
doi pubmed - Morgan GJ, Kelly JW. The kinetic stability of a full-length antibody light chain dimer determines whether endoproteolysis can release amyloidogenic variable domains. J Mol Biol. 2016;428(21):4280-4297.
doi pubmed - Kourelis TV, Dasari S, Theis JD, Ramirez-Alvarado M, Kurtin PJ, Gertz MA, Zeldenrust SR, et al. Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin light-chain amyloidosis by mass spectrometry. Blood. 2017;129(3):299-306.
doi pubmed - Jimenez-Zepeda V, Bril V, Lemieux-Blanchard E, Royal V, McCurdy A, Schwartz D, Davis MK. A comprehensive multidisciplinary diagnostic algorithm for the early and efficient detection of amyloidosis. Clin Lymphoma Myeloma Leuk. 2023;23(3):194-202.
doi pubmed - Vrana JA, Theis JD, Dasari S, Mereuta OM, Dispenzieri A, Zeldenrust SR, Gertz MA, et al. Clinical diagnosis and typing of systemic amyloidosis in subcutaneous fat aspirates by mass spectrometry-based proteomics. Haematologica. 2014;99(7):1239-1247.
doi pubmed - Lilleness B, Ruberg FL, Mussinelli R, Doros G, Sanchorawala V. Development and validation of a survival staging system incorporating BNP in patients with light chain amyloidosis. Blood. 2019;133(3):215-223.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Hematology is published by Elmer Press Inc.